
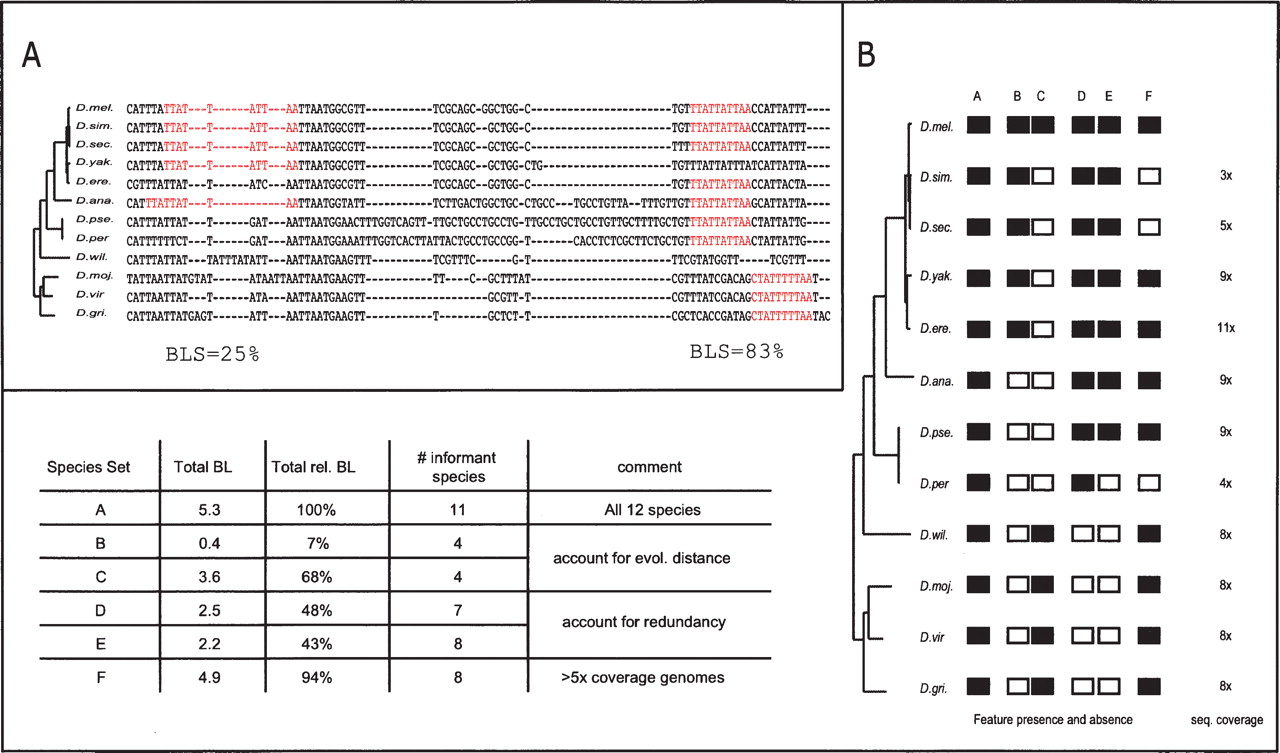
BLS measure (Branch Length Score) for assessing motif conservation in many genomes. (A) Conservation level and corresponding BLS scores for two Mef-2 motif instances. The BLS measure scores the total branch length of the subtree connecting the species with motif instances, as a fraction of the total branch length of all twelve species. As shown in these examples (Mef-2 motif: YTAWWWWTAR), BLS accounts for local alignment inaccuracies, gaps, motif movement, and motif loss. Species abbreviations as follows: Drosophila melanogaster (D. mel.), D. simulans (D. sim.), D. sechellia (D. sec.), D. yakuba (D. yak.), D. erecta (D. ere.), D. ananassae (D. ana.), D. pseudoobscura (D. pse.), D. persimilis (D. per.), D. willistonii (D. will.), D. mojavensis (D. moj.), D. virilis (D. vir.), and D. grimshawii (D. gri.). (B) BLS scores for different instance conservation scenarios. Given the pattern of presence (black) and absence (white) within a phylogenetic tree, BLS evaluates the total branch length of the subtree connecting the species that contain the motif: When all species are present, BLS is 100% (column A); different sets of species lead to different BLS scores based on their evolutionary distances: distantly related species lead to higher scores as they span larger evolutionary distances (columns B,C); species that are very closely related to each other lead to only small incremental contributions, due to their phylogenetic redundancy (columns D,E); sequencing, assembly, and alignment artifacts are not penalized, such as those stemming from lower-coverage genomes, as redundancy of branches between close species complements BLS (column F). Information about sequence coverage is from Drosophila 12 Genomes Consortium (2007) and Richards et al. (2005).








