
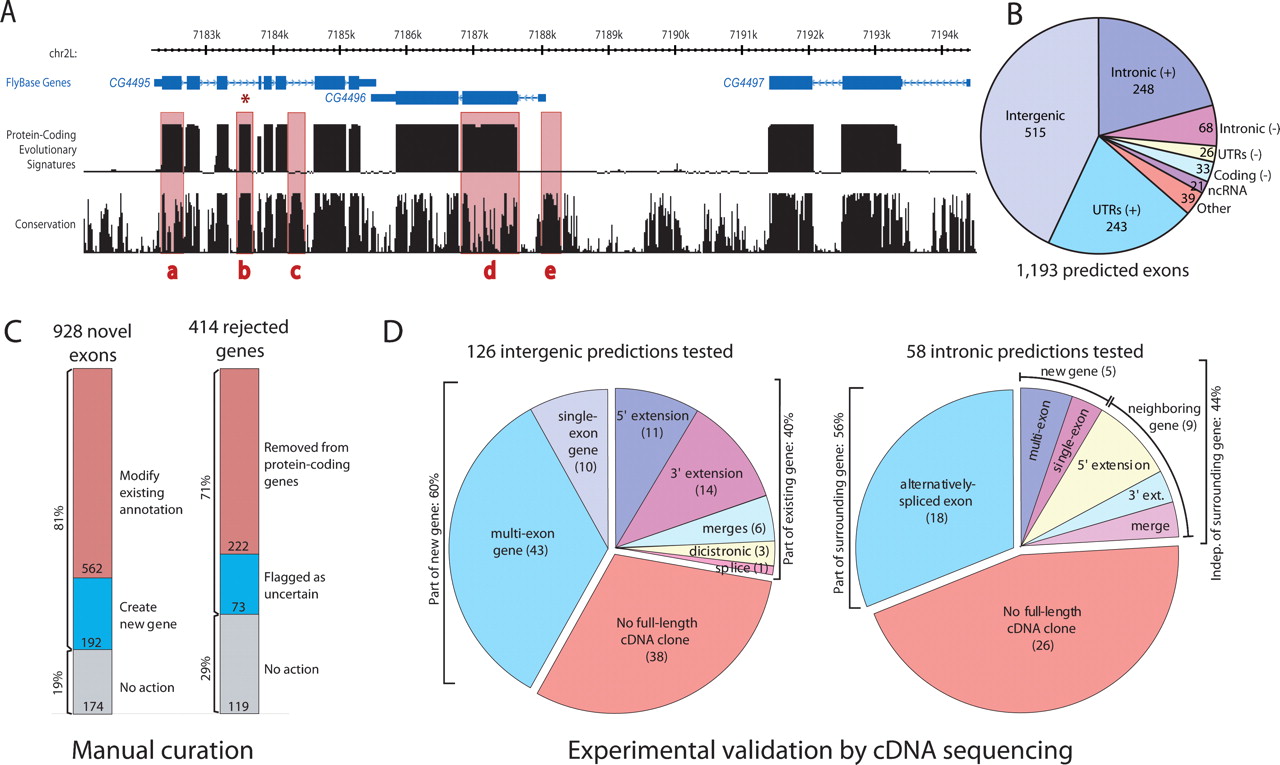
New protein-coding exons predicted by evolutionary signatures, examined by manual curation, and validated by cDNA sequencing. (A) The “Evolutionary Signatures” track shows the posterior probability of a protein-coding state in a probabilistic model integrating the RFC and CSF metrics. The “Conservation” track shows the analogous quantity from a model measuring nucleotide conservation only (Siepel et al. 2005). Note the high protein-coding scores of known exons despite lower nucleotide conservation (a,d), the low protein-coding scores of conserved noncoding regions (c,e), and the prediction of a novel exon within an intron of CG4495 (b), subsequently validated (see Fig. 3). Rendered by the UCSC Genome Browser (Kent et al. 2002). (B) Distribution of 1193 new exon predictions throughout the genome. (C) Newly predicted exons were examined by manual curation, 81% leading to new and modified FlyBase gene annotations. Additionally, curation of genes rejected by evolutionary signatures led to the recognition of hundreds of spurious annotations. (D) A sample of predicted new exons was tested by cDNA sequencing with inverse PCR. Surprisingly, 44% of the validated predictions in “intronic” regions revealed a transcript independent of the surrounding gene, and 40% of the validated predictions in “intergenic” regions were part of existing genes. See Fig. 3 for examples.








