
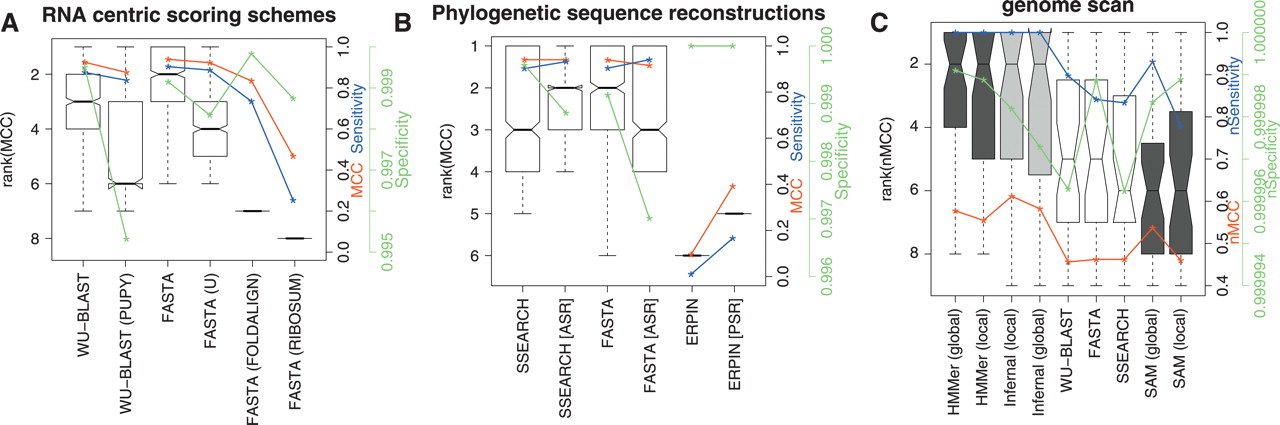
A comparison of the accuracy of methods using RNA-centric scoring matrices, phylogenetic sequence reconstructions, and the genome scan results. (A) A comparison of the accuracy of sequence-based methods with score matrices optimized for ncRNA. These boxplots show the distributions of the ranks on MCC for each of the homology search methods when using one of WU-BLAST (W7), WU-BLAST (W7, PUPY), FASTA, FASTA (U), FASTA (RIBOSUM), or FASTA (FOLDALIGN). These matrices are discussed in more detail in the text. (B) A comparison of FASTA, SSEARCH, and ERPIN with and without phylogenetic sequence reconstructions included in the input. Ancestral sequence reconstruction was used in the case of FASTA and SSEARCH and posterior predictive sequences in the case of ERPIN. Both A and B show results using 5 query sequences. (C) A set of representative programs from each category were run on human chromosome 12 (coordinates 90,000,000–130,000,000; ver NCBI35). The boxplot displays algorithm ranks; additionally, median nMCC, median nSensitivity, and median nSpecificity for each algorithm are displayed using the y-axis on the right. The single sequence, profile HMM, and RNA methods are displayed in unshaded, dark shaded, and lightly shaded boxes, respectively.








