
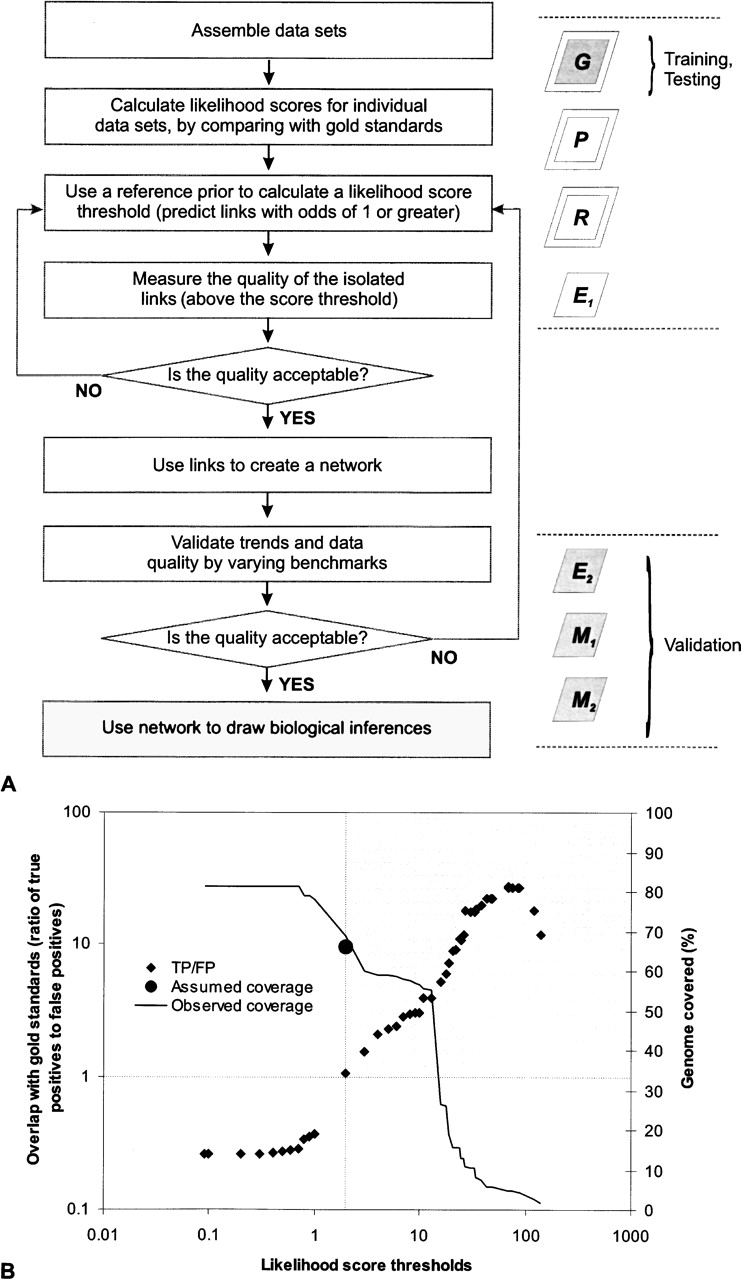
A generalized strategy for integrating diverse functional genomics data and reconstructing functional interaction networks. (A) A salient feature of this strategy involves the use of reference priors containing an assumed number of links, in the absence of knowledge about gene interactions in a genome. An appropriate prior that accurately reflects the state of genome can then be used to isolate a confident result set. (G) Benchmark gold standards; (P) phylogenetic profile data; (R) Rosetta stone linkage data; (E) gene expression data (E1: Bozdech et al. 2003; E2: Le Roch et al. 2003); (M) mass spectrometric data (M1 and M2). Dark boxes indicate benchmarks; boxes with double borders indicate in silico data. (B) Prior knowledge about the possible number of functionally interacting proteins leads to a corresponding likelihood score threshold, with posterior odds of finding a true pair of functionally linked proteins greater than one. Likelihood score thresholds and the ratio of true to false positives were calculated as described by Jansen et al. (2003). For our chosen threshold, the number of proteins assumed to functionally interact with each other corresponds very closely with the number of proteins with predicted functional linkages in our final data set. Linkages from the shaded area were used for network reconstruction. (Cross-hair) Chosen likelihood score threshold and estimated accuracy; (●) approximate coverage described by our prior belief; (♦) ratio of true positives to false positives.








