
Ultrasensitive RNA profiling: Counting single molecules on microarrays
The ability to analyze RNA expression of a whole genome in a single microarray experiment has had widespread impact on basic research as well as drug discovery and development (Marton et al. 1998; Brown and Botstein 1999; Bentwich et al. 2005). It also holds promise as a tool to guide treatment in the clinic (Golub et al. 1999; Perou et al. 1999; Alizadeh et al. 2000; Mattie et al. 2006; Yanaihara et al. 2006).
What else lies in the future for microarray technology? Until recently, researchers have rightly limited their horizons to what the technology can do rather than what it ought to do. However, there is agreement that it ought to be able to detect RNA from small amounts of sample material, even single cells, in a way that faithfully represents RNA abundances. In addition, there would be advantages to describing abundance levels in absolute terms— numbers or molar amounts—rather than relative values, so that comparisons between genes and across many experiments can be undertaken. Furthermore, the dynamic range of microarrays should match the range of expression levels found in cells (Holland 2002). Indeed, if the sensitivity, dynamic range, and quantitative nature of measurements could be improved, the current need for cross-validation with real-time PCR would become redundant.
In order to address these issues, a change in the way we look at molecules on a microarray is needed. At present, an ensemble signal is acquired from the plurality of labeled molecules that interact with probes in a microarray spot. However, if this signal were to be resolved into its constituent parts, the individual molecules, the output would be more easily quantitated because it would be digital: An individual molecule (one bit of information) can be either present or absent, the binary 1, 0. Moreover, if single molecules can be detected, then it follows that the detection is highly sensitive and the amount of sample material required can be reduced accordingly.
Although the detection of individually resolvable fluorescent molecules on surfaces has been described previously (Funatsu et al. 1995; Lizardi et al. 1998; Unger et al. 1999), analysis of microarrays at the single molecule level is more challenging, requiring the high-resolution scanning of centimeter-square areas with high speed. A recent study (Hesse et al. 2006) has shown the application of a fast CCD scanning method to a conventional long oligomer microarray spotted on a nonconventional slide, at a resolution that enables individual molecules to be resolved. Although individual Cy3 and Cy5 dye molecules—which are the labels most commonly used in microarray experiments—emit enough photons to be detected by a typical microarray scanner, scanners are not set up to resolve molecules individually. This is because of a trade-off between the time it takes to perform the scan and the resolution that is achieved. Conventional pixel-by-pixel scanners would take impossibly long (weeks) to scan 1 cm2 of a microarray at the ∼200-nm resolution required for single molecule analysis. Remarkably, the system used by Hesse et al. (2006) was able to scan a 1-cm2 area in under 1 h. This was done by a form of CCD operation, time-delay integration (TDI) or “scanning” mode, that is normally used in astronomy for finding trajectories of objects such as asteroids (Netten et al. 1994; Hesse et al. 2004). This technology synchronizes CCD read-off with a continuous stage movement. The imaging is done in strips, which are subsequently stitched together to reconstitute the microarray image. Simpler detection regimes could be implemented if brighter labels were used such as plasmon resonant nanoparticles (Oldenburg et al. 2002; Blab et al. 2006; also see Fig. 1A).
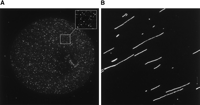
Single molecule detection and visualization. (A) A microarray spot containing oligonucleotide probes at low density hybridized with a complementary target labeled with 20-nm fluorescent nanoparticles. (Inset) Zoom view. (B) Genomic DNA molecules elongated on a slide surface.
The benefits of analyzing single molecules is clearly evident from Hesse et al.'s work. Without needing to use PCR or linear amplification, the Hesse group achieved a 100-fold decrease in the amount of sample material needed. This should open up applications where sample quantities are limiting. Also, the ability to work with small amounts of material without the need for amplification circumvents the preferential amplification of high-abundance messages such as globins in blood, which is one of the more accessible tissues for microarray analysis. Hesse et al. were able to validate their single molecule results by conventional microarray hybridization done with 100-fold more material. This is impressive, as different microarray platforms often do not show high concordance.
Hesse et al. (2006) also demonstrate that the dynamic range of single molecule detection is superior to conventional methods. The range of mRNA abundance levels in biological systems can approach 6 orders of magnitude, which clearly cannot be addressed by the ∼103 dynamic range of current microarray experiments. In contrast, the Hesse investigators found the dynamic range of their single molecule detection system to be 4.7 orders of magnitude. In particular, the range at the lower end, at which regulatory molecules may be expressed, can be extended with greater confidence. It is possible that by using an appropriate single molecule microarray system, the full 6 orders of magnitude of biological expression levels can be addressed in a single readout. One drawback of Hesse et al.'s implementation of single molecule detection on microarrays is that there is an upper transcript concentration limit. One of the three transcripts they studied in detail could not be analyzed at the single molecule level because its high abundance produced a density of binding events on the surface that, due to the diffraction limit of light, could not be resolved. In Hesse's system, the concentration of each transcript in the sample needs to be <10 fM for an optically resolvable signal density to be obtained. This would not be the case if the actual probe molecules within the microarray spots were placed at low density, on average beyond a diffraction-limited separation, so that no matter what concentration the target is, the density of hybridization signal is dictated by the density of the probe molecules (Fig. 2).
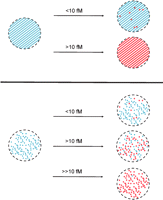
The effect of probe density and target concentration on single molecule detection on microarrays. Top panel illustrates the effect of hybridization of different target concentrations to a miroarray spot, containing probe molecules at normal spotting density (diagonal stripes). Single molecules can only be resolved when the labeled target molecule is below a threshold concentration (solid red circles); above the threshold, the density of signals is too high to detect single molecules. Bottom panel illustrates the effect of hybridization of different target concentrations to a single molecule microarray, containing probe molecules at a density low enough for single molecules to be resolved. Single molecules are resolved irrespective of the concentration of the labeled target; extension of the dynamic range is illustrated.
Hesse et al. (2006) noticed that the cDNA bound to the spot for one gene gave brighter signal peaks than a cDNA hybridized to the spot for another gene. This is not entirely unexpected because multiple labels are incorporated into strands during cDNA synthesis. Hence, single molecule cDNA foci will have signal brightness due to several dye molecules, and the occurrence of the nucleotide carrying the label may be less frequent in one cDNA than another. In conventional microarray analysis, this differential label incorporation would have given a higher signal in one spot than the other without it necessarily reflecting the difference in expression levels between the two genes. This confirms that researchers are right to limit their focus to gene expression fold changes rather than absolute levels when using conventional microarray formats. Clearly, the single molecule approach is able to provide more accurate quantitation, and together with other improvements (Rouse et al. 2004; Frigessi et al. 2005), an analysis based on single molecules is likely to be particularly appropriate for systems biology, where the assessment of comparative absolute expression levels is important for understanding the wiring of gene networks.
What Hesse et al. (2006) have demonstrated is impressive, but the benefits of taking microarray technology to the single molecule level do not stop there. Single molecule technology impacts the sensitivity issue not just by lowering the amount of sample material that is required but also in two other critical ways. Firstly, single molecule detection would enable the detection of the proverbial needle in a haystack. Because each molecule within the spot is assessed individually, a molecule present in only a few copies can be detected in the presence of messages in high copy number, which in conventional analysis would mask the less abundant message. This is particularly relevant for allele-specific transcript quantification. The second way that single molecule technology is sensitive is that very small differences in expression levels can be quantitated with confidence, without the extensive replicates and sophisticated statistical analysis needed to access small fold differences in conventional analysis (Pan et al. 2002; Wernisch et al. 2003).
The fact that single molecule technology is digital has a profound impact on the quality of data that are obtained. The intensity values obtained by conventional microarray methods are a composite of signal from the actual DNA interactions and contaminating fluorescence from artifacts, background signal from the slide glass, surface coating, instrument noise, and stray light. This contaminating light dampens the extent to which a signal stands out above background. In contrast, in the single molecule approach, sources of background noise other than nonspecific binding of DNA can be eliminated from the quantitation, and artifacts can be recognized and rejected. Only spatially distinct point sources of fluorescence characteristic of a binding event are counted, allowing extraction of a cleaner signal. The absence of noise leads to an increased range of linearity between the number of molecules in the sample and the quantitative measurement. Hesse et al.'s signal peaks stand >10-fold over background, enabling software to be devised to identify and count them; Oldenburg et al. (2002) showed an S/N ratio of 8.3 for counting compared to 3 for a conventional intensity analysis of the whole spot.
The features of single molecule analysis described for RNA profiling are also relevant for other types of biological or chemical analysis (Proll et al. 2006) requiring high sensitivity, wide dynamic range, and reliable quantitation. Moreover, the single molecule approach has other features that are relevant in applications beyond quantitation. Where a population of molecules need to be monitored over several events, the stochastic nature of molecular interactions leads to loss of synchrony between the molecules. In conventional ensemble analysis, this leads to a mixture of out-of-phase signals that do not allow the individual events to be resolved. This is relevant for studying biomolecular mechanisms, such as protein expression (Cai et al. 2006), and for some of the new sequencing methods that are under development (Shendure et al. 2004). In contrast, in the single molecule regime this is not a problem because each molecule can be tracked independently. A single molecule approach is also important when several features of the same molecule need to be assessed, for example, in molecular haplotyping (Woolley et al. 2000; Zhang et al. 2006). In this case, rather than visualizing single molecules as fluorescence point sources, they can be stretched out and visualized as long linear polymers (see Fig. 1B), upon which specific features can be mapped. It seems a wonder that, apart from a few exceptions (Herrick and Bensimon 1999), we continue to grapple in the dark with indirect inferences—such as the relationship between migration of a DNA molecule on a gel and its length—rather than shining light directly on the stuff of life.
Footnotes
-
E-mail kalim{at}well.ox.ac.uk; fax 44-1865-287533.
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.5825506.
- Copyright © 2006, Cold Spring Harbor Laboratory Press








