
Structure and function of the human genome
Abstract
The human genome project has had an impact on both biological research and its political organization; this review focuses primarily on the scientific novelty that has emerged from the project but also touches on its political dimensions. The project has generated both anticipated and novel information; in the later category are the description of the unusual distribution of genes, the prevalence of non-protein-coding genes, and the extraordinary evolutionary conservation of some regions of the genome. The applications of the sequence data are just starting to be felt in basic, rather than therapeutic, biomedical research and in the vibrant human origins and variation debates. The political impact of the project is in the unprecedented extent to which directed funding programs have emerged as drivers of basic research and the organization of the multidisciplinary groups that are needed to utilize the human DNA sequence.
The past decade in biological research has surely been the decade of genome research—from the scientific perspective, in the public imagination, and even in the minds of international politicians. It is therefore timely to use this 10th anniversary of Genome Research to take stock of where we are and where we might be in another decade in our understanding of the human genome.
The scale of the human DNA sequence must mean that no reviewer can capture all of the information it contains and therefore I concentrate on what novel information emerges from the completed sequence rather than on the detail of what we learned from each gene or each base.
The Human Genome Project (HGP) has had scientific and political impacts on biological research; scientifically, it has provided a novel conceptual dimension to human biology, that of “completeness.” This word captures the idea that we now have finite bounds to research because the genome sequence contains all of the information that is used in making human cells and organisms. We can soon legitimately claim to study the behavior of all of our genes in a way that was quite inconceivable prior to the availability of the sequence. Politically, the HGP is changing our perspectives on how biological research can be organized in our institutions. This review inevitably focuses on the scientific outcomes, but toward the end of the review, I discuss the idea that perhaps the HGP's significant long-term impact will be on the organization of scientific research.
The original inception of the HGP included optimistic views of the impact of knowledge of our genome on biomedical research (see, e.g., Collins et al. 1998), and the first biomedical impacts of the HGP are fundamental insights rather than pharmaceutical outcomes. For example, sequence analysis has led to the identification of new oncogenes (for review, see Strausberg et al. 2004), and microRNA composition is being used as a novel classifier of human tumors (He et al. 2005; Lu et al. 2005), but such information is presently distant from therapeutic outcome. The lack of immediate application of HGP data is unsurprising given the >10-yr drug development pipeline (Dickson and Gagnon 2004). Over the next decade we will see an accumulation of basic knowledge derived from the genome sequence, and this will then inform therapeutics, suggesting that benefit must necessarily be deferred.
If the biomedical goals of the HGP are in the future, the immediate outcomes expected by the scientific community were perhaps more pragmatic; a description of the gross structure of the human DNA sequence, the number of genes, and the proteins these might encode. Along with these reasonable expectations was the hope that the primary DNA sequence would reveal clues as to the control of gene expression. Secondary outcomes included describing the sequence variation between humans and, closely related to this, insights into the evolutionary and population history of our genome.
The present (assembly number 35, May 2004) human DNA sequence contains ∼3,100,000,000 bp (depending on the actual source of the assembled DNA sequence) that covers most of the nonheterochromatic portions of the genome and contains some 250 gaps (see Fig. 1). Its analysis has produced both predictable and novel insights. In the predictable category are the complete description of base compositional bias, the variation of rates of recombination in relation to the physical DNA length, the high proportion of the genome comprising repetitive DNA sequences, and, more ambivalently, the identification of many genes of known and unknown function (Venter et al. 2001; International Human Genome Sequencing Consortium 2004). Essentially, in these areas, the HGP has simply extended what we already knew without adding wholly novel insight. In contrast, and the primary focus of this review, unexpected insights are being gained from the identification and analysis of genes and their distribution, the amount of transcription of non-protein-coding regions, and the large-scale duplication structure of the genome.
Genes in the human genome
Perhaps the most publicly discussed result of the HGP was the realization that we have ∼20,000-25,000 genes (International Human Genome Sequencing Consortium 2004), somewhat fewer than estimates based on the preliminary reports of the human sequence (International Human Genome Sequencing Consortium 2001; Venter et al. 2001). Identifying genes—the process known as “annotation”—has predominantly been achieved through bioinformatics, most particularly by homology analyses and some de novo gene predictions. These data are readily accessible through several large genome “browsers” (for review, see Karolchik et al. 2003; Birney et al. 2004). The recent detailed analysis of 1% of the human genome under the ENCODE (ENCyclopedia Of DNA Elements) project (ENCODE Project Consortium 2004; http://www.genome.gov/10005107) indicates that these approaches have a relatively high success rate at identifying the presence of a gene within a region but a much lower success in predicting the gene's structure correctly (see, e.g., Brent and Guigo 2004); this suggests that annotation may underestimate gene number but not substantially. The low gene number prompted press comment on the difficulty of equating human complexity with apparent genetic simplicity; such comment seems to ignore the extraordinary combinatorial possibilities that can be generated from the interaction of even small numbers of gene products, a fact noted well before the final figure had been released (Ewing and Green 2000).
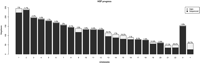
The sequenced (gray filled) and unsequenced (white) portions of the human genome, listed by chromosome; numbers in % are the proportion of chromosomes that are heterochromatic and unsequenced for this reason. Statistics are from the NCBI Build 35, UCSC assembly of May 2004, Assembly 17, data from http://www.genome.ucsc.edu/goldenPath/stats.html#hg17.
The extensive annotation process also confirmed the importance of alternative splicing in creating proteome diversity. Presently, estimates for the per gene frequency of alternative splicing range from 35% to ∼60% (Johnson et al. 2003), but there remains substantial uncertainty in determining the extent to which these estimates reflect functionally significant splices or splice errors (for review, see Sorek et al. 2004). The influence of alternative splicing on proteome complexity (for review, see Southan 2004) is a matter of substantial biological importance, and lack of precision in predicting genes, gene structures, and alternative splices necessarily limits the present utility of genomic information; these are areas that must see substantial direct experimentation before a nearly complete data set can emerge.
Non-protein-coding RNA transcripts: The relationship of genes and transcribed regions
In parallel with the low gene number, there is accumulating evidence that there are many transcripts that appear to be non-protein-coding and of no known function (Cheng et al. 2005; Kapranov et al. 2005; for review, see Johnson et al. 2005), an observation that is mirrored in the mouse (for review, see Suzuki and Hayashizaki 2004). In humans, the original observations were controversial both because the level of RNA produced from these so-called transfrags (transcribed fragments) can be low and also because transfrags are often not annotated as genes; both concerns prompted doubts about the biological importance of such transcription. There are several reasons that these concerns may be unnecessary. J. Manak and T. Gingeras (pers. comm.) have shown that in early development in Drosophila, many of these transfrags are, in fact, alternative unannotated 5′ start sites of otherwise annotated genes. If this finding is true for humans, it is tempting to believe that the transcription may be involved, for example, in reorganizing a chromatin domain so that it can subsequently be transcribed in a controlled fashion later in development. Secondly, these transfrags necessarily sequester RNA polymerase and relevant accessory proteins, and it is possible that the biological relevance of transcription might simply be in relation to the control of availability of the basal and cell-specific transcription factors. These speculations are as yet untested.
There has also been considerable speculation that noncoding RNAs might have a regulatory function, and in part these proposals have been influenced by the increasing evidence that the DNA of many genes is transcribed off both coding and noncoding strands (see, e.g., Kapranov et al. 2005). An essential role for some noncoding RNA transcripts in early embryonic development had been demonstrated by transgenesis long before the more general analysis of the genome (Brunkow and Tilghman 1991), and the role of antisense transcripts in regulating human genes is well documented (for recent review, see O'Neill 2005). The challenge of studying the function of the many new examples of antisense and noncoding transcripts is considerable, since it will require sophisticated manipulation of relevant regions to establish likely function; some of these analyses may emerge from the ENCODE project discussed below (ENCODE Project Consortium 2004).
MicroRNAs are a class of noncoding RNA that are the focus of increasing attention since their initial description in animals (see, e.g., Lagos-Quintana et al. 2001). The number of human microRNA genes in the genome may be >800 (Bentwich et al. 2005), and a significant majority of these are of unknown function. The increasing data that support a fundamental role for this class of noncoding RNAs (see, e.g., He et al. 2005; Lu et al. 2005) are driving research in this area, and the next few years will see progressively clearer descriptions of the number and biological role of these RNAs.
Emerging from these data is the realization that our concept of a gene is becoming somewhat unclear at a molecular level. In particular, the relationship of transcription to gene expression, to control of gene expression, and even to control of translation has become more complex, and by this measure a greater proportion of the genome is functional than we previously understood. It is important to recognize that function in these cases is being used in two different senses; in one extreme use, function resides entirely in the specific DNA sequence of a region (e.g., a transcription factor binding site), but at the other extreme, “structural” function can be quite independent of sequence (e.g., spacer DNAs). This view has critical implications for interpreting patterns of sequence conservation that show that overall only ∼5% of our sequence is subject to selective evolutionary pressure and therefore “functional” (for review, see Miller et al. 2004).
The distribution of genes within DNA
The gene distribution in the full sequence provided two surprises: firstly, striking gene-poor “deserts”; regions of up to 3 Mb (Venter et al. 2001) that are devoid of genes, with a statistically high probability that these are not the tails of a random distribution of genes. In the mouse, deletion of two deserts had no immediate phenotypic consequences (Nobrega et al. 2004). Presently, there is no satisfactory explanation for the existence of gene deserts, but the varying pattern of conservation within deserts suggest some function; Nobrega et al. 2003, Ovcharenko et al. (2005), and de la Calle-Mustienes et al. (2005) show that some deserts contain enhancers distant to flanking genes.
Secondly, prior to the results of the HGP, the location of genes along the DNA was known to be functionally important; clusters of coordinately expressed genes such as the HOX or globin clusters were well studied, but it was clear that these clusters were products of gene duplication events in deep evolutionary time. However, Yamashita et al. (2004) identified large-scale functional clustering of genes that were coexpressed in specific human tissues. Boon et al. (2004) and Petkov et al. (2005) reported similar results in the mouse, and Caron et al. (2001) reported clustering of genes expressed at high levels into specific chromosomal regions. Importantly, the clusters do not appear to be the products of evolutionary duplications of an ancestral gene(s), and the implication is that clustering reflects some level of coordinate control, speculatively, such as enhancer sharing or open chromatin conformation.
Elements that control gene expression
The identification of cis-acting promoter sequences that control gene expression has inevitably become the focus both of intensive bioinformatics analysis (see, e.g., Liu and States 2002 or Zhang 2003) and experimental research (Kim et al. 2005). Perhaps the most difficult aspect of bioinformatics predictions is testing the results in practical experimentation, and here the ENCODE project is a key development. Presently, the ENCODE project has the goal “to identify all functional elements in the human genome sequence” (ENCODE Project Consortium 2004; http://www.genome.gov/10005107) by using a mix of different direct experimental and computational approaches. The challenge of these studies is considerable; many promoters function bidirectionally (Trinklein et al. 2004), and the relationship of transcription to “gene” expression is, as noted above, becoming more complex.
It is here that we can perhaps predict the next significant development of the HGP as a collaborative project because we face a severe technical and biological challenge—technically because evidence to date suggests that no one approach to elucidating gene control is satisfactory, and biologically because the tissue specificity of gene expression requires us to study its control, ultimately, in all human tissues. To meet these challenges is a task that will require coordination; perhaps systematic genome research (as opposed to research using genome information) should initially be concentrated on multiple technical approaches, targeted at a collaboratively agreed small number of well-studied cell types. Ideally, these should include the genetically well-characterized CEPH lymphoblastoid cell lines that have been extensively characterized for genetic variation in the Human Haplotype Map (the “HapMap”) project (International HapMap Consortium 2003; http://www.hapmap.org). Such a project would certainly synergize cellular biological, genetic, and clinical studies to an unprecedented extent.
Arguably, one of the most surprising results of the HGP was the identification (Bejerano et al. 2004; Siepel et al. 2005) of regions of the genome, called “ultra conserved elements” (UCEs), that were extraordinarily highly conserved between evolutionarily distant species. The human genome contains 481 such regions that are >200 bp in length (see Fig. 2) and are 100% invariant between the human, rat, and mouse sequences. This conservation is far greater than can be accounted for by protein-coding constraints of an absolutely conserved protein or by requirements of RNA secondary structure. Recently S. Salama and D. Haussler (pers. comm.) have shown that some UCEs are enhancer elements of nearby genes, and this suggests a potential solution to the puzzle of their ultraconserved nature. Enhancers contain multiple transcription-factor-binding sites, and any given factor can bind to a family of short DNA sequences consisting of a mix of highly invariant or relatively unconstrained bases (Transfac database at http://www.gene-regulation.com/pub/databases.html). A testable hypothesis to explain the extraordinary conservation of some UCEs is to suggest that they consist of clusters of transcription-factor-binding sites organized as partially overlapping sets, such that the invariance of a base in one binding site defines the identity of an otherwise variable base in a second partially overlapping factor-binding site. The overall result of the overlap of factor-binding sites would be a DNA sequence that could not be altered, since variation of a base would disrupt the function of one or more transcription factors; such a sequence would therefore be highly resistant to evolutionary change.
Large-scale structures in DNA
The sequence revealed the full extent to which human DNA is comprised of abundant interspersed repeats, extending and completing what was already known; fully 45% of our DNA consists of repetitive elements interspersed within nonrepetitive sequences. Interestingly, the extent and diversity of gene repetitions contained in low copy number repeats were greater than expected; very extensive duplications of regions of DNA both within and between chromosomes were identified by the International Human Genome Sequencing Consortium (2001) and Venter et al. (2001).
Some years prior to the HGP and based on the identification of genes in multiples of four in our DNA, the suggestion was made that the human genome was a quadrupalized derivative of a smaller ancestral genome (see, e.g., Spring 1997). Analysis of the complete sequence fails to support this hypothesis, because there is no significant increase in fourfold repeated regions in the genome.
More recently She et al. (2004) have extended the initial analyses to define the full duplication landscape of the genome, and Tuzun et al. (2005) have shown that there are significant copy number polymorphisms between individuals, the phenotypic consequences of which in many cases are unknown. The location of deletions, insertions, and inversions are shown in Figure 3.

The location of 481 Ultra conserved elements (UCEs) in the human genome with a detailed display of the UCEs within the POLA gene. Reproduced with permission from Science © 2004, Bejarano et al. (2004).
Human genetic variation
The Human Haplotype Map (the “HapMap”) project is a key component of realizing the genetic potential of the HGP (The International HapMap Consortium 2003; http://www.hapmap.org). This project is based on identifying DNA sequence variations, predominantly single nucleotide polymorphisms (SNPs), in a target of 270, ethnically diverse human beings. The SNPs are grouped into haplotypes to provide a descriptive framework on which human phenotypic (and further genotypic) variation can be mapped. The extent to which haplotypes capture human variation is still unclear (see, e.g., Evans and Cardon 2005; Sawyer et al. 2005), and we are still far from having reasonable estimates of how explicitly haplotypic variation influences, or is correlated with, phenotypic variation. The potential for the HapMap to inform analysis of human complex genetic disorders was one of the founding principles of the project, and the next five years will see the full application of this research. More conjecturally, the reduced complexity of haplotype sharing between two individuals, when compared to the full sequence difference, may allow us to introduce wholly novel genotypic classifications of human diversity. Such a development would have an important impact on population- and cohort-based research such as clinical trials and on the genetic basis of personalized medicine.
Recreating human ancestry
It was always understood that the HGP would provide the framework for the study of human diversity from a biomedical perspective but that these data could equally be applied to the study of human history through tracing historical patterns of migration and population structure. We can certainly anticipate that the HapMap will provide an enormous intellectual platform to power these analyses; the present expense of genotyping by resequencing will necessarily limit the extent to which human populations may be studied. There is every reason to suppose that the driver of biomedical research will force sequencing costs down to levels where large-scale study of populations by resequencing will become cost tractable; the outcome will be the most detailed description of human origins that these technologies and human history can allow.
Human ancestry can also be studied by comparison with the great ape DNA sequences, and this is an emerging area of research that has captured imaginations widely, particularly in respect to the evolution of human higher cognitive functions such as language (Enard et al. 2002). The difficulty of these studies is that the divergence of human and chimpanzee DNA at ∼1.23% is so small (The International Chimpanzee Chromosome 22 Consortium 2004; Chimpanzee Sequencing and Analysis Consortium 2005) that it does not easily allow statistically robust identification of selection, and our understanding of the genetic basis for higher cognitive function remains sketchy.

Location of 139 insertions, 102 deletions, and 56 inversions on each human chromosome, showing the location positioned against the DNA sequence. Reproduced and modified with permission from Nature Genetics © 2005, Tuzun et al. (2005).
The role of comparative sequence analysis in annotation has been amply demonstrated in the HGP and in many other genome projects. A novel use of this information allowed Blanchette et al. (2004) to attempt to recreate the molecular genetic ancestors of humans and other vertebrates (see Fig. 4). Conventional DNA sequence phylogenetic analyses use statistical approaches to establish a likely order of changes to DNA in the course of evolution and thus recreate an evolutionary history. Blanchette et al. (2004) used the ∼1.8-Mb CFTR gene region DNA sequence from 18 mammalian species, but instead of focusing on the order of changes, they attempted to recreate the ancestral DNA sequence by statistical modeling to “reverse” base changes to the evolutionary basal state—creating the eutherian ancestral genome (Fig. 4). That there are statistical limitations to this approach is certainly recognized, but perhaps in the not-too-distant future, we may be able to combine our theoretical knowledge of ancestral DNA sequences with our knowledge of evolutionary developmental biology to put flesh on the “bones” of the DNA sequence of an unknowable distant common ancestor!
Broadening the impact of the HGP
So far this review has focused on the achievements of the HGP in terms of novel information and concepts generated from within the project itself, but, of course, one of the founding principles underlying the HGP was that the DNA sequence would inform a very wide range of research. Has this goal been achieved? It is clear that even the incomplete knowledge of the genic and therefore protein composition of humans has, indeed, supported much research but has not, perhaps, produced the flood of new therapies and concepts that more enthusiastic supporters had proclaimed.
Can the impact be broadened? Completing the annotation of genes would certainly contribute to increased impact by facilitating the technical exploitation of genome information, for example, enhancing our ability to define canonical DNA probes on microarrays, and contributing to biological study of human genes. Of course, the ethical limitations of research on humans has restricted the scope of experimental descriptions of tissue specificity of gene expression and of alternative splice forms. Human embryogenesis is a particularly difficult area of study, and it is likely that human embryonic (and other) stem cells will become very important surrogate targets for experimental analysis of the human gene complement and its control.
The recreation of an ancestral DNA sequence. This is an example based upon a MER20 retrotransposon. The sequence labeled “PRED.ANC.” at the bottom is the prediction of the ancestral MER20 sequence. Reproduced and modified with permission from Cold Spring Harbor Laboratory Press © 2004, Blanchette et al. (2004).
The human sequence display—genome browsers
The HGP data is exceptionally rich in information but most critically, this richness is very much in the eye of the beholder; the genome sequence holds very different information for different biologists. For this reason, the second area where the HGP impact may be widened is in the difficult task of presenting the DNA sequence for use by the scientific community. In the introduction to “A User's Guide to the Human Genome” (Wolfsberg et al. 2003), it was noted that “many investigators whose research programs stand to benefit in a tangible way from the availability of this information have not been able to capitalize on its potential.” In part, as the guide tried to argue, this was because of user unfamiliarity with the complex data available through the normal browsers that display the sequence, those of the UCSC (http://genome.ucsc.edu/; Kent et al. 2005), NCBI (http://www.ncbi.nlm.nih.gov/genome/guide/human/; Wheeler et al. 2005), and Ensembl (http://www.ensembl.org; Hubbard et al. 2005). However a potentially more significant factor to be considered is that these interfaces are all designed around the view of the DNA sequence as the ultimate genetic map, which is a geneticist/genomicist view of the genome. Most experimental biologists are not interested in genetic organization but rather are interested in biological organization—for example, proteins expressed or functioning in the same space or same time in an organism. The descriptive language of gene function is becoming formalized around the terms defined in the Genome Ontology project (The Gene Ontology Consortium 2000; http://www.geneontology.org), and this provides an important framework for description. Displays based on this more complex knowledge are presently intended for more specialist users (e.g., the proteomics community), and until they become more generic, it is likely biologists will remain somewhat distant from genome information. The next decade will most assuredly see enormous strides in this area, particularly under the integrative drive of systems-biology-based approaches.
The organizational lead of the HGP
The HGP was one of the few examples in biology of an attempt to coordinate and focus research to specific goals by a strongly directed program of investigation. The HapMap and the Encode projects are good examples of the second generation of directed projects, and while both may have their scientific critics, both are clearly generating novel information that will support much hypothesis-driven research in the future. A continuing role for directed research is ultimately a political decision of some complexity if only because of the mix of individual creativity and more collective endeavor that is characteristic of the best of genome research. In any event, the HGP has in many respects led the way in modern biological research; it has altered the politics of science funding very substantially by virtue of its scale, of the scale of the information that biologists can now access and of the complexity of that information. Genomic-scale analyses have started to revolutionize biological research, drawing computer scientist, mathematician, biologist, clinician, chemist, and physicist into complex collaborative projects. This is surely one of the realized beneficial outcomes of the HGP, realized far in advance of the impact of increased biological knowledge itself. I would argue that one of the achievements of the HGP has been to alter the way in which we study ourselves, and this, it seems to me, is as profound an impact as one can hope for in a field as complex as human biology.
Conclusions
I have focused on novelty in this review, but the undeniable reality is that the human sequence presently, and for decades to come, underpins an extraordinary range of research that ultimately is only limited by the interests of those who use its information. This is the real success of the HGP, but it is a success that does not readily lend itself to headlines. Genome information does not allow us to escape from the extraordinary complexity of our biology, and thus it is not a golden source of drugs, drug targets, cures, and insights. Its information cannot be read like a book because that is not the logic of living cells. Post-genome sequence, science is quite unlike anything we have previously encountered, but the brutal reality is that our own biology remains as difficult to study as it has ever been; perhaps, therefore, the greatest contribution the HGP has made is to show us just how complex we really are.
Acknowledgments
Space limitations do not allow me to cite many of the relevant references in this review; I hope the individuals concerned can forgive the necessary omissions! I am indebted to the past and present members of my lab and colleagues for their help in forming my understanding of genome biology: this work was supported by a grant from the Australian ARC to P.F.R.L. and through the expertise of The Clive and Vera Ramaciotti Centre for Gene Function Analysis at the University of New South Wales.
Footnotes
-
E-mail p.little{at}unsw.edu.au; fax 61-2-9385-1483.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.4560905.
- Cold Spring Harbor Laboratory Press








