
Advances in livestock genomics: Opening the barn door
Abstract
Genome research in animals used in agriculture has progressed rapidly in recent years, moving from rudimentary genome maps to trait maps to gene discovery. These advances are the result of animal genome projects following closely in the footsteps of the Human Genome Project, which has opened the door to genome research in farm animals. In return, genome research in livestock species is contributing to our understanding of chromosome evolution and to informing the human genome. Enhancement of these contributions plus the much anticipated application of DNA-based tools to animal health and production can be expected as livestock genomics enters its sequencing era.
The Human Genome Project is properly credited with accelerating the discovery of disease genes and providing a totally new paradigm for medical research. It has sharpened our approach to the study of development, behavior, cancer, and infectious diseases, and provided a toolbox for deciphering genetic diversity and human origins. It probably does not get enough credit, however, for opening the door for genetic analysis of other animals, particularly those species used in agriculture. Livestock genomics has followed in the footsteps of the human genome initiative, adopting both its successful strategies and technologies to advance our understanding of livestock genomes with shoestring budgets relative to the resources available for human and medical research. In turn, livestock genomics contributes to informing the human genome. Mapping and sequencing species from clades other than primates and rodents contribute to our understanding of evolutionary history and its underlying mechanisms. As demonstrated by Thomas et al. (2003) and others, sequences from these diverse clades contribute to the identification of functional elements in the human genome outside the more easily annotated coding regions.
In addition to informing the human genome, agricultural science has a unique responsibility to human health and social stability, and that is feeding an expanding world population while minimizing environmental and ecological risks. Clearly, the identification of variation in livestock genomes that predisposes health and productivity with less reliance on hormones, antibiotics, and pesticides will be a major step in meeting this global challenge. A review of recent advances in livestock genomics and their realized and potential contributions to both human biology and agricultural science is the goal of this paper.
One of the world's most important agricultural animals and one in which genomics research advanced rapidly to the sequencing phase, is the domestic chicken. That genome is the subject of another review in this issue (Burt 2005), however, and this review focuses on the mammals, principally cattle, pigs, sheep, and horses, and to a lesser extent, river buffalo and goats.
Status of livestock genomics
Early attempts to construct whole-genome maps of livestock species were based on the two technologies underlying the first human genome maps, somatic cell genetics and in situ hybridization (Womack and Moll 1986; Yerle et al. 1995). These early maps defined synteny (genes on the same chromosome but not necessarily linked) and cytogenetic locations of sequences hybridizing specific DNA probes. These strategies proved extremely important to early comparative mapping because the mapped markers were generally genes or gene products, highly conserved across mammalian genomes. The synteny and cytogenetic maps they produced gave us our first insights into the relative stability of the mammalian genome throughout its evolutionary history and pointed to certain groups of animals with a much higher degree of genome similarity than others, suggesting some lineages of animals with highly conserved genomes and others in which genomic evolution was much more dynamic. Linkage mapping, however, lagged behind, awaiting the development of highly polymorphic markers with sufficient density in the genomes of outbred animal populations to efficiently map traits with whole-genome approaches. Beckmann and Soller (1983), inspired by advances in human genetics, were early proponents of the use of DNA level markers for building maps and mapping traits in livestock species. Modern genomics in livestock followed the lead of the human genome initiative at all levels and had its formal origins in a series of conferences in which strategies were distilled, and more importantly, collaborations were established to maximize the relatively meager resources available to animal genetics in the early 1990s. Two international conferences in 1990—a Banbury Conference on “Mapping the Genomes of Agriculturally Important Animals” at Cold Spring Harbor and the first Allerton Conference in Illinois on “Gene Mapping of Domestic Animal Genomes: Needs and Opportunities”—were springboards for collaborative use of available genomic resources and the prioritization and development of resources unavailable at that time. From these and other similar gatherings, international groups of animal geneticists launched both formal and informal genome projects for some of the most widely used livestock species, resulting in our current inventory of genomic resources (Table 1). Following is a brief discussion of the development and current status of genomic information for cattle, pigs, sheep, horses, and the lesser developed genomes of river buffalo and goats. Recent comprehensive reviews focused on individual species are available for cattle (Lewin 2003; Sonstegard and Van Tassell 2004), pig (Rothschild 2003), sheep (Cockett 2003), horse (Chowdhary and Bailey 2003), buffalo (Iannuzzi et al. 2003), and goat (Schibler et al. 1998). Moreover, Andersson and Georges (2004) reviewed livestock genomics in the context of grand challenges of human genomics (Collins et al. 2003), providing both a model for this paper and a standard for measuring the development of livestock genomics in its role of helping to define the human genome through the genetic analysis of complex traits.
Map status and genomic resource for livestock species
Cattle
Cattle genomics had its origins in somatic cell genetics (Heuertz and Hors-Cayla 1981; Womack and Moll 1986; Womack 1987). The first “genome maps” for cattle were synteny groups, genes on the same chromosome, defined by protein gene products segregating in hybrid somatic cell lines. These synteny groups were assigned to specific chromosomes by integrating somatic cell genetics with in situ hybridization (Fries et al. 1986, 1993; Gallagher Jr. et al. 1993) and were greatly expanded with the advent of molecular markers, initially defined by probed Southern blots and later by PCR-based markers. An international consortium organized at the 1988 meeting of the International Society for Animal Genetics (ISAG) assembled a set of families for linkage mapping, and the development of microsatellite markers in the early 1990s resulted in an international linkage map (Barendse et al. 1994), almost concurrently with a linkage map developed at USDA-MARC (Bishop et al. 1994). These maps were quickly expanded (Barendse et al. 1997; Kappes et al. 1997) into tools that have proved effective for mapping loci underlying both monogenic and quantitative traits. The next significant advance in cattle genomics was the development of radiation hybrid (RH) maps (Womack et al. 1997; Williams et al. 2002) and the use of these maps for high-resolution comparative mapping (Band et al. 2000; Everts-van der Wind et al. 2004; Itoh et al. 2005). A consortium to generate a bacterial artificial chromosome (BAC) map of the bovine genome has generated a 294,651 whole clone HindIII fingerprint map that is currently being refined by BAC end sequencing and is scheduled for completion in 2005. Highly developed linkage and RH maps and the progress of the BAC consortium were instrumental in the success of a White Paper proposal to the NHGRI for whole-genome sequencing in cattle (http://www.genome.gov/Pages/Research/Sequencing/SeqProposals/BovineSEQ.pdf). A single partially inbred Hereford female was selected to contribute 6× whole-genome shotgun (WGS) reads and another 1.5× will come from individual animals of the Holstein, Angus, Jersey, Limousin, Brahman, and Norwegian Red breeds for SNP detection. In addition, an ∼1× BAC skim sequence will aid assembly. Sixfold WGS has been achieved and breed skims for SNP detection are under way. Approximately 10,000 nonredundant full-length cDNAs are being sequenced at the Michael Smith Genome Science Center in Vancouver. The University of Illinois has generated a 3800-element bovine cDNA microarray (Band et al. 2002), recently upgraded to 7872 elements (Everts et al. 2005), and The Bovine Functional Genomics Consortium (Suchyta et al. 2003) has produced an 18,000-element array.
Pigs
The history of pig genomics does not parallel that of cattle in that the linkage map was the first whole-genome map produced. As a result of the European PiGMaP initiative (Archibald et al. 1995) and USDA-MARC (Rohrer et al. 1996), linkage maps were developed and subsequently expanded to a high level of resolving power for trait mapping. An estimated 2700 markers are represented on these two maps. Somatic cell genetics (Yerle et al. 1996) and RH mapping (Yerle et al. 1998; Hawken et al. 1999) have facilitated comparative mapping in pigs. As in cattle, RH mapping of ESTs with human orthologs has provided the power for definitive comparative mapping of the pig and human genomes (Rink et al. 2002; Tuggle et al. 2003). An international group has begun construction of a BAC map (http://www.genomic.iastate.edu/newsletter/PigWhitePaper.html, NHGRI), and large cDNA sequencing and EST projects are advancing rapidly (Fahrenkrug et al. 2002; Tuggle et al. 2003). Multinational funding has recently been secured for whole-genome sequencing to begin in 2005. Meanwhile, the “Sino-Danish Pig Genome Project” has published pig genome sequence with <1× coverage (Wernersson et al. 2005). Functional genomics in pigs is now the beneficiary of a 3468-element microarray (Niewold et al. 2005) and a 3867-element microarray (Dvorak et al. 2005) both from intestinal mucosa.
Sheep
Burkin et al. (1993) developed a somatic cell hybrid panel segregating sheep chromosomes and assigned a few dozen markers to syntenic groups before somatic cell genetics in sheep gave way to linkage mapping. Building on linkage maps developed to find the Booroola fecundity gene, Montgomery et al. (1993) and Crawford et al. (1995) produced a map with 246 markers. Interestingly, approximately one-half of these markers were bovine-derived microsatellites. Second-generation (de Gortari et al. 1998) and third-generation (Maddox et al. 2001) linkage maps have been expanded to include well over 1000 markers. Radiation hybrid maps were developed at INRA (France) and Utah State University (Cockett 2003) and are currently being populated with both EST and microsatellite markers, a strategy that integrates genetic and physical maps. BAC libraries have been produced and contigs assembled around several regions of interest to individual laboratories.
Horse
Horse genomics was slow out of the gate but has picked up its pace significantly in the last five years. A workshop in 1995 (http://www.uky.edu/AG/Horsemap/) launched an international initiative that has produced three linkage maps (Lindgren et al. 1998; Guérin et al. 1999, 2003; Swinburne et al. 2000) totaling in excess of 450 markers, more than 400 of which are microsatellites. A comprehensive RH map was produced by Chowdhary et al. (2002) that contains 730 markers and integrates somatic cell, linkage, and cytogenetic maps into a valuable tool for comparative mapping (Chowdhary and Bailey 2003). Radiation hybrid mapping of the horse X-chromosome revealed perfectly conserved gene order relative to the human X-chromosome at a moderate level of RH resolution (Raudsepp et al. 2002).
Buffalo
The world population of river buffalo used for meat and milk consists of >130,000,000 animals. Scientific resources are limited in many of the countries where buffalo are economically important livestock, and as a consequence genome research has not been supported at the level of some of the other species. Excellent cytogenetics and fluorescence in situ hybridization, however, principally in the laboratory of Leopoldo Iannuzzi, has established a strong foundation for buffalo genomics. Almost 300 loci are on the cytogenetic map (Iannuzzi et al. 2003), most of them with homologs mapped in other species, and thus have contributed significantly to comparative mapping. The development of a hybrid somatic cell panel (El Nahas et al. 1996) produced synteny maps that were integrated with cytogenetic maps, resulting in the immediate assignment of syntenic groups to chromosomes. Chromosome banding patterns revealed almost identical karyotypes of river buffalo (2N = 50) and cattle (2N = 60) with five bi-armed buffalo chromosomes appearing to be fusions of five pairs of single-armed cattle chromosomes. Identity of these chromosome arms has been verified by comparative mapping in the two bovid species. A recently developed radiation hybrid panel for buffalo (E. Amaral, J. Elliott, J.E. Womack, pers. comm.) will advance comparative mapping to a higher level of resolution. There are presently no linkage maps for river buffalo. The work of E. Amaral, J. Elliott, J.E. Womack (pers. comm.), however, suggests that as in sheep, primers for most cattle-derived microsatellites amplify buffalo sequences in homologous regions of the respective genomes. If sufficient numbers of these microsatellites are polymorphic in buffalo, they will facilitate the development of a linkage map when pedigreed families are properly identified and DNA is made available to the growing buffalo mapping community.
Goat
A linkage map of the goat (Vaiman et al. 1996) was expanded significantly by Schibler et al. (1998) and now contains >300 markers. The latter study also added 202 cytogenetic localizations, many of which were also mapped by linkage, thus integrating the two maps. This map was among the first integrated maps in ruminants and served as a prototypic ruminant map and an excellent tool for comparative mapping prior to the generation of radiation hybrid maps in cattle and sheep.
Contributions of livestock species to comparative mapping
Along with that of the domestic cat (O'Brien and Nash 1982), genome maps of livestock species were the first to expand the comparative maps of mammalian genomes beyond primates and rodents. Early somatic cell maps were largely driven by comparative mapping interest; thus homologs of genes previously mapped, usually in humans, were genotyped in panels of hybrid somatic cells derived from domestic animals. Markers segregating together in these panels were said to be syntenic or on the same strand (chromosome). The term “synteny” is in and of itself irrelevant to comparative genomics and was coined by Frank Ruddle to describe genes determined to be on the same chromosome by somatic cell genetics as opposed to the term “linkage,” which has traditionally been associated with nonrandom assortment of alleles of two or more genes in meiosis. Unfortunately, the term synteny is often adulterated to imply evolutionary conservation of homologous chromosome segments between species. Nonetheless, the first autosomal comparative maps in non-rodent species were comparisons of syntenic groups in humans to syntenic groups in domestic animals (O'Brien and Nash 1982; Womack and Moll 1986). These were subsequently aided by cytogenetic mapping of homologous genes in different species.
Comparison of syntenic groups between two species is a type of comparative chromosomal painting if the markers are ordered in one species. Developing unordered synteny maps in cattle, for example, using ordered homologous markers from the human map results in the equivalent of “cattle on human” painting of the human map. The opposite, human on cow (or pig, horse, etc.) painting, was made possible by the technique of Zoo-FISH (Wienberg et al. 1982; Jauch et al. 1992). The hybridization of cocktails of fluorescence labeled unique sequence probes derived from isolated human chromosomes to chromosomes of another species was first applied to non-human primates but subsequently to other species including cattle (Hayes 1995; Solinas-Toldo et al. 1995; Chowdhary et al. 1996), pigs (Goureau et al. 1996), and horses (Raudsepp et al. 1996). Single chromosome paints are not generally available from the domestic animal species for Zoo-FISH painting with the exception of pig (Goureau et al. 1996).
As mentioned earlier, comparative genomics at the level of DNA sequence is particularly instructive in the identification of highly conserved genomic elements other than coding sequences (Margulies et al. 2003; Thomas et al. 2003). The power of this approach has been enhanced by the availability of whole-genome sequence from the livestock species. An exciting harvest from comparative genomics will come when multiple species from different evolutionary clades have been sequenced or mapped at a high level of resolution. Highly conserved elements in cattle, sheep, goat, and buffalo, for example, might point toward what makes a ruminant a ruminant if conserved homologs are not obvious in other clades. Similar comparisons could point to unique functional elements in primates, rodents, and other groups with multiple sequenced species.
A significant contribution to biology from farm animal genomes is evident in the recent discovery of extensive reuse of chromosome breakpoints during mammalian evolution (Murphy et al. 2005). These sites of evolutionary activity are marked by high gene density, accumulation of segmental duplications in humans, and footprints of telomeres and centromeres. Similar studies will undoubtedly continue to uncover biologically significant sites in animal genomes that are not revealed by concentrated focus on a single genome.
Informing human medicine
The livestock species have some distinct advantages over other animals for studying the underlying mechanisms of phenotypic variation between and within species. Yet, the farm animals have not been fully appreciated or exploited in biomedical research. All the livestock species were domesticated from wild ancestors in the last 10,000 years, and highly differentiated phenotypes have resulted from intensive selective breeding, much of it in the last century. In addition, large numbers of offspring can be produced from a single mating in most species, and excellent phenotypic records accompany many of the pedigrees of animals with extreme phenotypes. And finally, highly developed genomic tools and rapidly developing databases are now available for the study of the major domestic animal genomes.
Biological insights gained from animal genomics include aiding the discovery of genes for human diseases. An excellent example is the discovery of a mutation in the MC4R gene in pigs (Kim et al. 2000) that results in obesity similar to that in humans. A good example of gene discovery in animals leading directly to gene discovery in humans is a mutation in the limbin gene responsible for chondrodysplastic dwarfism in Japanese Brown cattle (Takeda et al. 2002). Limbin had not previously been associated with any of the inherited dwarfisms in humans but was subsequently determined to be the homolog of EVC2, a gene responsible for Ellis-van Creneld syndrome, an autosomal recessive chondrodyplastic dwarfism in humans (Galdzicka et al. 2002; Ruiz-Perez et al. 2003). Discovery of the cattle gene clearly aided the discovery of the gene underlying the human disease.
Double muscling, a generalized muscular hypertrophy in cattle, has been recognized for almost 200 years and has been positively selected in some breeds such as the Belgian Blue, where the recessive mh allele is practically fixed. Conversely, it is an undesirable phenotype in many breeding programs because of the high incidence of dystocia. The gene was mapped to bovine chromosome 2 by Charlier et al. (1995) and fine-mapped by “identity by descent” (IBD) by Dunner et al. (1997). Comparative mapping (Dunner et al. 1997; Sonstegard et al. 1997) placed the mutation in a region of cattle chromosome 2 that is conserved relative to human chromosome 2. Comparative candidate positional cloning (Womack 1996) suggested several candidate genes from the human map, including myostatin (Grobet et al. 1997; Smith et al. 1997). Meanwhile, McPherron et al. (1997) demonstrated pronounced muscular hypertrophy in mice homozygous for a knockout deletion of Gdf8, the locus encoding myostatin. Based on this strong comparative genomic information, Grobet et al. (1997) sought and found a deletion in the bovine myostatin gene responsible for the ms/ms phenotype. Other loss-of-function mutations in the bovine gene have been identified in Belgian Blue and other breeds (Kambadur et al. 1997). Schuelke et al. (2004) recently described a child with gross muscular hypertrophy. The wealth of literature on double muscling in cattle and the mouse knockout experiments immediately led these investigators to the human myostatin gene, where, indeed, a splice-site mutation was discovered. Thus, loss of function of the myostatin gene produces similar phenotypes across the three mammalian species (Fig. 1), and the identification of mutations in double-muscled cattle was instrumental in discovery of the homologous gene underlying the similar human phenotype. Various methods of blocking myostatin function are being considered as therapies for muscular-degenerative disorders in humans (Bogdanovich et al. 2002).
Improving animal health and production
Selection for desirable traits, or conversely, selection against undesirable traits, has been practiced since the domestication of animals almost 10,000 years ago. The promise of more accuracy, efficiency, and economy in selecting animals that will produce offspring with desirable phenotypes, however, underpins a substantial portion of the funding for livestock genome projects over the past two decades. The early linkage maps for most livestock species were constructed as tools for mapping traits and developing molecular markers for use in marker-assisted selection (MAS). The ultimate marker for MAS is, of course, the mutation underlying the selected phenotype. Although mapped QTLs in livestock species now number in the hundreds, very few mutations underlying quantitative trait variation have been identified. For obvious reasons, success has been better with several monogenic traits of economic and biological interest. In an extremely valuable database called “Online Mendelian Inheritance in Animals (OMIA)” (http://www.angis.org.au/oma/), Frank Nicholas lists 56 single-locus traits in cattle, 27 of which have had the causative mutation identified. The same listing provides equivalent numbers of 33 and 11 for pig, 59 and 9 for sheep, 26 and 9 for horse, and 8 and 5 for goat. Thus, the mutation has been identified in only about one-third of the single-gene traits cataloged in these species. Of the hundreds of QTLs now mapped in livestock, we have only two examples of the elucidation of mutations underpinning the QTL, both in dairy cattle. The first discovery of a quantitative trait nucleotide (QTN) was provided by comparative candidate positional cloning of the DGAT1 locus as a gene contributing to fat composition in milk on chromosome 14 (Grisart et al. 2002), followed by functional confirmation of the effect of a mis-sense mutation (Grisart et al. 2004). Another QTL for milk fat and protein concentration was identified on chromosome 6 (Ron et al. 2001), and recently localized to a mis-sense mutation in the ABCG2 gene, again with the aid of comparative and functional analysis (Cohen-Zinder et al. 2005).
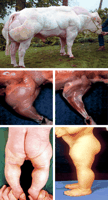
Phenotypic comparisons of: (top) “double muscled” bull homozygous for loss-of-function allele at the myostatin (MSTN) locus. Photograph courtesy of Michel Georges, University of Liège, Belgium. (Middle) Forelimb of GDF-8 (myostatin) knockout mouse (left) versus wild-type littermate (right). Photograph courtesy of Se-Jin Lee, Johns Hopkins University School of Medicine, Baltimore. Reprinted with copyright permission from Nature Publishing Group © 1997, from McPherron et al. 1997 (http://www.nature.com/). (Lower) Child at age of 6 d (left) and 7mo(right) homozygous for loss-of-function allele in the myostatin gene. Photograph courtesy of Markus Schuelke, University Medical Center, Berlin. Reprinted with copyright permission from the Massachusetts Medical Society and the New England Journal of Medicine © 2004, from Schuelke et al. 2004.
Although QTN discovery has been slow in all mammals, including humans, a promising future was predicted by Korstanje and Paigen (2002) with their charting of exponential growth of genes and mutations identified in mammalian QTL studies beginning in 1999. These discoveries parallel the development of genome sequencing initiatives. The emergence of sequence from livestock species over the next few years bodes well for the discovery of genes underlying health and production traits in economically important species.
The not too distant future
My public statements in the late 1990s that domestic animal genomes would likely never be fully sequenced disqualify me as a prognosticator in the dynamic discipline of livestock genomics. Nonetheless, it seems inappropriate to conclude a review of our discipline without a brief guess, albeit a conservative one, as to where it might be headed. Genome sequencing, database development, expression arrays, and SNP maps with automated genotyping will obviously become staples of our genomic toolbox, probably before our current generation of graduate students leaves our laboratories. RNA interference may soon find its way into animal improvement, likely in conjunction with cloning from modified somatic cells. It is interesting to speculate on the early applications of these genomic resources. While the bottleneck between mapped QTL and gene discovery will not be cleared immediately, we should expect a rapidly accelerated harvest of causative mutations for biologically interesting and economically important phenotypes. The next wave of livestock QTLs will likely lead us to the discovery of new genes for disease resistance. Selection of animals with innate resistance to pathogens is, of course, important to sustainable agriculture and one potential defense against agricultural bioterrorism. With few exceptions such as the discovery of the role of BoLA-DBR3 in bovine leucosis (Xu et al. 1993) and mapping of the QTLs for trypanotolerance in cattle (Hanotte et al. 2003), variation in disease resistance has been recalcitrant to either candidate gene studies or QTL mapping in livestock species. This generally reflects our inability to safely contain pathogens in challenge experiments requiring large numbers of cows, pigs, sheep, and the like. Gene sequencing and SNP discovery in our domestic animal species will soon give us information about linkage disequilibrium over large genomic regions and the identification of haplotype blocks in various populations and breeds of livestock. It is likely that these blocks will be large enough in many populations for retroactive association studies in herds subjected to pathogen exposure. The list of molecules involved in host recognition of pathogens and associated cell signaling grows almost daily, suggesting ample opportunity for mutations throughout the genome to differentiate the response of individual animals to pathogen contact. The identification of QTLs for disease resistance in livestock may be the next big frontier for the contribution of domestic animal genomics to the understanding of host–pathogen interaction and the subsequent improvement of both animal and human health.
Footnotes
-
[The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: M. Georges, S.-J. Lee, M. Schuelke.]
-
E-mail jwomack{at}cvm.tamu.edu; fax (979) 845-9972.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3809105.
- Cold Spring Harbor Laboratory Press








