
Insights on biology and evolution from microbial genome sequencing
Abstract
No field of research has embraced and applied genomic technology more than the field of microbiology. Comparative analysis of nearly 300 microbial species has demonstrated that the microbial genome is a dynamic entity shaped by multiple forces. Microbial genomics has provided a foundation for a broad range of applications, from understanding basic biological processes, host-pathogen interactions, and protein-protein interactions, to discovering DNA variations that can be used in genotyping or forensic analyses, the design of novel antimicrobial compounds and vaccines, and the engineering of microbes for industrial applications. Most recently, metagenomics approaches are allowing us to begin to probe complex microbial communities for the first time, and they hold great promise in helping to unravel the relationships between microbial species.
During the past 10 years, genomics-based approaches have had a profound impact on the field of microbiology and our understanding of microbial species. Since the first report on the complete genome sequence of Haemophilus influenzae in 1995 (Fleischmann et al. 1995), nearly 300 other prokaryotic genome sequences have been completed (http://www.genomesonline.org/; http://cmr.tigr.org/tigr-scripts/CMR/CmrHomePage.cgi), with another 750 projects underway. In the early days of microbial genomics, our ignorance about the extent of species diversity was reflected in the assumption that the complete sequence of 20-30 carefully chosen representatives of the bacterial and archaeal domains of life would provide a sufficient amount of information for follow-up investigations. As sequence data began to accumulate, it quickly became clear that we had underestimated the wealth of genetic and biochemical diversity in the prokaryotic world. Indeed, the completion of the sequence of Escherichia coli O157:H7 by Perna and colleagues in 2001 revealed that this new isolate contained more than 1300 strain-specific genes as compared with E. coli K-12. These genes encode proteins involved in virulence and expanded metabolic capabilities, as well as several prophages. This was a striking example of the fact that two members of the same species could differ in gene content by almost 30%. Today, many genomics efforts are focused on sequencing multiple isolates and strains and providing new insights into species diversity and the dynamic nature of the prokaryotic genome.
Because of their larger genome sizes, genome sequencing efforts on fungi and unicellular eukaryotes were slower to get started than projects focused on prokaryotes; however, today there are a number of genome sequences available from both of these groups of organisms that have led to significant improvements in overall sequence annotation and also shed considerable light on novel aspects of their biology (see Dolinski and Botstein 2005; Galagan et al. 2005).
A changing view of the microbial world
The microbial world can be classified into four groups that differ in many aspects of their biology: Bacteria and Archaea, which represent the prokaryotes, single-celled Eukarya, and viruses. During the past 10 years, a large and phylogenetically diverse number of microbial species has been targeted for genome analysis. Extremes in genome size (<500 kb to almost 10 Mbp) and gene content have also been revealed by these studies, with no absolute boundaries between viral, bacterial, archaeal, fungal, and protist genomes (Fig. 1).
When one considers the more than 20-fold difference in bacterial genome size (Fig. 1), a question that emerged is whether or not one can define a minimal set of genes essential for life. The notion of a minimal genome has been explored through a number of both experimental (Hutchison III et al. 1999; Sassetti et al. 2003; Krause and Balish 2004) and theoretical (Koonin 2003; Klasson and Andersson 2004) approaches. One of the first studies employed transposon mutagenesis in the minimal organisms Mycoplasma genitalium and Mycoplasma pneumoniae, based on the assumption that there would be a limited number of genes encoding proteins with redundant functions, thereby making it easier to identify essential versus non-essential genes. The results of this study suggested that as many as 130 of the 480 predicted coding sequences may not be essential in vitro (Hutchison III et al. 1999). However, one limitation of the transposon approach is that the mutants that are generated contain disruptions only in single genes. To date, there has been no experimental validation that a minimal genome containing 350 functional genes would support life. Indeed, successive rounds of M. genitalium mutagenesis revealed that fitness of the cultures is gradually reduced (Peterson and Fraser 2001). While it might be possible to compensate for reduced fitness through gene loss by supplementation of the growth medium, this notion points out the intricate relationship between the definition of a minimal genome and the cellular environment. More recently, new methods have been described for the deletion of large, non-essential regions of the E. coli genome (Kolisnychenko et al. 2002; Goryshin et al. 2003; Hashimoto et al. 2005) and are allowing for correlations between genotype and phenotype. While these techniques show great promise for engineering reduced bacterial genomes, other new approaches for generating 5- to 6-kb segments of DNA from oligonucleotides (Smith et al. 2003) and for microchip-based multiplex gene synthesis (Tian et al. 2004) represent a significant step toward an era of synthetic biology that will also enable some of the current hypotheses about minimal gene sets to be tested experimentally.
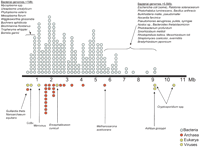
Depiction of overlapping genome size in members of the Bacteria (blue), Archaea (red), Eukarya (green), and viruses (yellow), in the size range (∼0.5-10.5 Mb) in which this overlap has been found to occur. Number of circles at a given point on the scale indicates the number of completed genomes of a specific size. Circles representing unusually small (<1 Mb) or large (>5.5 Mb) bacterial genomes are labeled with the species name. Reprinted with permission from Elsevier © 2005, from Ward and Fraser 2005.
Multiple forces are shaping microbial genomes
Comparative genomics approaches have revealed that the prokaryotic genome is a dynamic entity, different in many respects from more stable multicellular eukaryotic genomes. Multiple forces have shaped the prokaryotic genome during its evolution; these include gene loss/genome reduction, genome rearrangement, expansion of functional capabilities through gene duplication, and acquisition of functional capabilities through lateral gene transfer (Fig. 2).
Genome projects on various obligate intracellular pathogens and endosymbionts have provided several windows on the process of reductive evolution (Andersson and Kurland 1998; Moran and Plague 2004). Although these organisms are similar in that they contain significantly reduced genomes (≤1 Mbp) that are missing one or more key metabolic pathways, thereby making them dependent on their hosts for survival, it is clear that there are multiple solutions to a minimal genome. Evidence has suggested that the transition to obligate intracellular species often involves deletions of large segments of DNA early on in the process, likely catalyzed by genome instability mediated by insertion sequence (IS) elements or other mobile DNA that are eventually lost from the genomes (Moran and Mira 2001; Moran and Plague 2004; Belda et al. 2005; see also the example below from comparative analysis of Burkholderia spp). While it was initially thought that genes targeted for loss during this process were ones that were no longer necessary for survival in a host environment, there are increasing numbers of examples from comparative genomics projects that suggest some gene loss may be beneficial to the microbe. This is true in the case of certain pathogens, including Mycobacterium tuberculosis (Tsolaki et al. 2004), which has lost metabolic pathways and become more virulent, and Shigella (Nakata et al. 1993) and Bordetella pertussis (Parkhill et al. 2003), which have lost genes encoding cell surface antigens, a situation that may allow for enhanced ability to evade the host immune system.
Genome rearrangements mediated by IS elements can also play a major role in genome plasticity. The extent of such rearrangements often reflects the lifestyle of the organism. In general, obligate intracellular organisms that exist in relative isolation contain few mobile elements, and their genomes tend to be stable over long periods of time. At the other extreme, free-living bacteria often contain large numbers of IS elements and repetitive DNA sequences that may mediate homologous recombination. One example of the role of IS elements in mediating genome-wide rearrangements comes from the comparative analysis of Burkholderia mallei (Nierman et al. 2004), an obligate mammalian pathogen that causes glanders, and Burkholderia pseudomallei (Holden et al. 2004), an environmental soil-dwelling organism. The B. mallei genome contains 171 complete or partial IS elements that collectively represent >3% of the sequence (Nierman et al. 2004). The B. mallei genome is 1.4 Mb smaller than that of its closest relative, B. pseudomallei, and most of the synteny break points between the two genomes are bounded by IS elements. In addition, two syntenic portions of chromosome 1 in B. pseudomallei that are found on chromosome 2 in B. mallei are flanked by IS elements, lending further support to the idea that genome rearrangement can play a large role in genome structural alteration in certain species (Nierman et al. 2004).
Gene duplication and functional diversification is yet another mechanism for generating diversity within microbial genomes. Gene paralogs (genes related by duplication) can represent as much as 50% of the larger microbial genomes, and an interesting subset of such genes are lineage-specific duplications that presumably are responsible, in part, for species-specific biology. A recent analysis of 115 completed prokaryotic genome sequences by Konstantinidis and Tiedje (2004) using the Clusters of Orthologous Groups (COGs) database (Tatusov et al. 2003) revealed that larger genomes are disproportionately enriched in genes encoding proteins involved in regulation, secondary metabolism, and transport. The inverse correlation was observed with proteins involved in translation and DNA processing. This analysis provides a possible explanation for why species with larger genomes are more apt to dominate environments where nutrients are scare, because they are more versatile in terms of their ability to sense and respond to changing environmental conditions.
Another source of genome variability that plays an important role in prokaryotic genome evolution is lateral gene transfer (LGT), which brings new genes into a genome and provides a means for rapid adaptations to changing demands on an organism (Boucher et al. 2003). For example, acquisition of virulence determinants on pathogenicity islands appears to play a major role in pathogen evolution. Phylogenetic analysis of multiple strains of Staphylococcus aureus indicated that the diversification of the highly variable RD13 region, encoding putative pathogenesis-related proteins, has likely occurred by LGT and recombination (Fitzgerald et al. 2003). Comparative analysis between S. aureus and its close relative, Staphylococcus epidermidis, demonstrated that genome islands in non-syntenic regions of the genome, likely acquired by LGT, are the primary source of variations in pathogenicity and virulence between these species (Gill et al. 2005). The acquisition of a circular plasmid with 99.6% similarity with the Bacillus anthracis toxin-encoding plasmid, pXO1, by Bacillus cereus G9241 was likely responsible for the emergence of a strain of B. cereus able to cause a disease resembling inhalation anthrax (Hoffmaster et al. 2004). Although homologs of the B. anthracis pXO2-encoded capsule genes were not found in this strain of B. cereus, a polysaccharide capsule cluster encoded on a second, previously unidentified plasmid, pBC218, was identified. The virulence of this stain was confirmed in an A/J mouse model of anthrax. LGT probably also plays an important role in generating diversity among environmental bacteria. Several lines of evidence have suggested extensive LGT from archaeal species to the hyperthermophilic bacteria, Thermotoga maritima (Nelson et al. 1999; Nesbo et al. 2002; Mongodin et al. 2005).
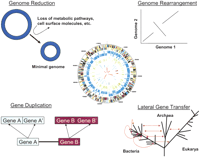
Multiple forces, including genome reduction, genome rearrangement, gene duplication, and acquisition of new genes via lateral gene transfer, are shaping microbial genomes. Details of each of these processes can be found in the text.
As prokaryotic genome sequences began to accumulate, there were a number of attempts to generate whole-genome phylogenies; however, these often resulted in trees that were incongruent with phylogenies based on rRNA and suggested that it may be difficult, if not impossible, to reconstruct the Tree of Life given the extent of LGT (Doolittle 1999). While there has been considerable debate about the frequency of LGT (Gogarten et al. 2002; Lawrence and Hendrickson 2003), the source of laterally transferred genes (Daubin et al. 2003a), the most robust methods for detecting LGT (Koonin et al. 2001; Ragan 2001; Snel et al. 2002), and its impact on phylogeny (Bapteste et al. 2004), more recent analyses have suggested that it is possible to extract a coherent phylogenetic pattern from analysis of a “core” set of genes (Daubin et al. 2003b; Phillipe and Douady 2003). While a more detailed discussion of LGT is beyond the scope of this review, there appears to be a consensus that it is perhaps most appropriate that the evolution of prokaryotic species is best depicted as a network of vertically and laterally transferred genes, rather than as an single tree. A recent report by Kunin and colleagues (2005) has suggested that certain organisms may serve as hubs for rapid LGT across species. One implication of this hypothesis is that a gene(s) conferring a selective advantage may traverse across many species with a small number of LGT events.
Given the extent of LGT that has been described in numerous studies, a question that can be posed is whether or not it is possible to define the pan-genome for any given bacterial species, that is, the total number of genes associated with all strains of an organism. A recent study by Tettelin and colleagues (2005) that examined diversity in eight isolates of Streptococcus agalactiae has suggested that the number of genes associated with this species may be theoretically unlimited. The pan-genome can be divided into three parts: a core genome shared by all strains, a set of dispensable genes shared by some but not all isolates, and a set of strain-specific genes associated with each isolate examined. The core genome encodes basic aspects of S. agalactiae biology, while the dispensable and strain-specific genes contribute to the genetic diversity of the species and the ability to colonize certain niches. This contrasts with B. anthracis, for which the pan-genome can adequately be described by four genome sequences. This difference in the type of pan-genome may reflect several factors, including the different lifestyles of the two organisms (i.e., exposure of S. agalactiae to diverse environments vs. occupation of a more isolated biological niche by B. anthracis), the ability of each species to acquire and stably incorporate foreign DNA, and an advantage in niche adaptation by acquisition of laterally transferred DNA.
Comparative genomics of unicellular eukayotes has also come of age with the completion of genome sequencing projects on a range of organisms, including a number of apicomplexa (Plasmodium spp., Theileria spp., Cyrptosporidium spp.), trypanosomes (Trypanosoma brucei, T. cruzi, and Leishmania major), amoebae (Dictyostelium discoideum and Entamoeba histolytica), microsporidia (Encephalitozoon cuniculi), and nucleomorphs (Guillarida theta). It is of interest that there are a number of parallels between unicellular eukayotes and prokaryotes. As observed with bacterial species, these unicellular eukaryotes differ tremendously in genome size, genome organization (chromosome number, gene density, and the presence and size of introns), and gene number. Genome reduction is also a force in the unicellular eukaryotic world, as evidenced by the minimal genomes of G. theta (0.55 Mb) (Douglas et al. 2001) and E. cuniculi (2.51 Mb) (Katinka et al. 2001). In these species, gene density, at approximately one gene per 1.2 Mb, approaches that observed in the prokaryotic world, and many metabolic genes have been lost, making these species dependent on their hosts for energy and small molecules. Other unicellular eukaryotes, such as two species of Theileria (Gardner et al. 2005; Pain et al. 2005) and Cryptosporidium (Abrahamsen et al. 2004; Xu et al. 2004) protists, represent organisms with moderately compact genomes with a gene density of approximately one gene per 2 Mb. In these cases, genome reduction also results from gene loss, particularly with regard to metabolic genes and plastid-related genes (Keeling 2004), together with reduced intron content (Cryptosporidium) or reduced intergenic space (Theileria). Despite the fact that a number of unicellular genomes have undergone reductive evolution, recent studies on two Cryptosporidium spp. (Abrahamsen et al. 2004; Xu et al. 2004) and the protist Entameoeba histolytica (Loftus et al. 2005) have lent support to the idea of unicellular eukaryote acquisition of bacterial genes involved in cellular metabolism through LGT.
Applications of microbial genomics
Approximately 40% of the bacterial species that have been targeted for genome analysis represent important human pathogens. Comparative in silico methods are allowing for correlations to be made between genotype and phenotype in many instances. For example, the Chlamydiaceae represent a group of closely related obligate intracellular pathogens that cause a range of diseases in mammalian and avian hosts. Genome analysis of several members of this clade has revealed a limited number of variable metabolic and cell surface genes, clustered in the replication termination region, that account for much of the differences in tissue and host tropism between species (Read et al. 2003; Thomson et al. 2005). An unexpected finding from Chlamydia genome sequencing projects was the discovery of a number of genes with similarity to enterobacterial virulence factors (Read et al. 2003), suggesting that the Chlamydiae may have been reservoirs for virulence genes at some distant point in the evolution of the enterobacteria.
Transcriptomic and proteomic approaches have also provided insights into genes involved in virulence, the molecular basis of host specificity, and host-pathogen interactions. One advantage of such large-scale approaches is the ability to monitor global changes in gene and protein expression in both the pathogen and the host during the infectious process. Another is that they can be used to study genes and proteins whose function is unknown. Recent transcriptome studies of Neisseria meningitidis—a causative agent of septicemia and meningococcal meningitis—provide an excellent example of how transcriptome analysis can be exploited (Grifantini et al. 2002; Dietrich et al. 2003). These studies showed that there were distinct sets of genes that were differentially regulated during two key steps in the meningococcal infection of human cells—the initial interaction with epithelial cells in the respiratory tract, and the later interaction with endothelial cells in the blood-brain barrier. These differentially regulated genes—which encode membrane transporters, transcription factors, general metabolic pathways proteins, and a number of hypothetical proteins—are obvious candidates for further studies that in turn could lead to novel approaches to preventing diseases caused by N. meningitidis.
One of the goals of genome-enabled research on human pathogens is the development of novel diagnostics, antimicrobial compounds, and vaccines. While progress is being made on all fronts, there have been a number of successes in the application of genome sequence data to the identification of novel vaccine candidates. A new method, reverse vaccinology, has been described that allows for identification of potential vaccine candidates based on genomic information, rather than the more traditional approach toward vaccine development pioneered by Pasteur more than two centuries ago, which requires growing the infectious agent as a first step (Rappuoli 2000). This approach has been successfully applied to rapid vaccine development against a number of human pathogens, including Neisseria meningitidis (Pizza et al. 2000; Tettelin et al. 2000), Streptococcus pneumoniae (Ross et al. 2001; Wizemann et al. 2001), Chlamydia spp. (Grandi 2003), as well as the viral pathogen that causes severe acute respiratory disease (SARS) (Bukreyev et al. 2004; Yang et al. 2004). While reverse vaccinology has proven to be very promising in generating a protective immune response in various animal models of disease, the results of ongoing clinical trials of these novel vaccine candidates will provide the ultimate test of the effectiveness of this approach to find new vaccines of benefit to humans.
Because of their unique metabolic properties, a variety of environmental organisms with potential utility in catabolic degradation of toxic compounds or other bioremediation processes have also been targeted for sequencing and functional analysis. As one example, Geobacter sulfurreducens is a member of the δ-proteobacteria and has the ability to precipitate soluble metals such as iron and uranium as a by-product of electron transport. Genome analysis of G. sulfurreducens (Methe et al. 2003) revealed the presence of a large number of c-type cytochromes and suggested the existence of a large number of redundant electron transport networks. A subsequent study demonstrated that electron transport in G. sulfurreducens occurs through direct contact of the pili with iron oxides and suggests that it may be possible to engineer biologically based conductive materials (Reguera et al. 2005). Continued exploration of the metabolic capabilities of microbes with potential bioremediation and biotransformation capabilities will be facilitated by the increasing availability of genome sequence data, together with the development of tools and databases for reconstruction of metabolic pathways such as EcoCyc (Keseler et al. 2005), the Biocatalysis/Biodegradation Database (Hou et al. 2004), and OptStrain (Pharkya et al. 2004), and new approaches for gene and genome synthesis (Smith et al. 2003; Tian et al. 2004).
Metabolic engineering of microbes is an area of long-standing industrial interest, especially for the production of small molecules. Genomics-based methodologies, including comparative DNA sequencing, transcriptome, proteome, and metabolome profiling, together with in silico modeling and simulation, have become important tools in bioengineering strategies (for review, see Lee et al. 2005). The potential of this approach to generate predictive models of cellular behavior was recently demonstrated by Fong et al. (2005), who engineered E. coli strains with improved production of lactic acid based on the genome-scale metabolic analysis together with adapted evolution of the new strains. The use of “omics” data to generate predictive models is still in its infancy and is limited by several factors, including a comprehensive lack of information on regulatory networks and the challenges of integrating data across multiple scales and times. However, one of the most exciting areas of future genome-enabled research will be in systems biology.

Applications and future directions in microbial genomics. The availability of microbial DNA sequence has provided a new foundation for follow-up studies, both in vitro and in silico. The ultimate goal is to integrate data from these multiple approaches to achieve a new systems-level understanding of the microbial cell.
Metagenomics: Another new frontier
Despite all of the progress in microbial genomics in the past 10 years, it is important to remember that essentially all of the projects completed to date have been focused on species that can be grown in culture. Given that >99% of the prokaryotes in the environment cannot be cultured in the laboratory, we are still greatly limited in our knowledge about the physiology and ecology of microbial communities (Schloss and Handelsman 2005). While small-subunit ribosomal RNA (rRNA) genes have been used in surveys of diversity in the uncultured prokaryotic world for some time, this information cannot provide any insights into the biology or species interactions in communities. The more recent application of high-throughput sequencing technology together with newer algorithms for sequence assembly have given rise to the field of metagenomics (Riesenfeld et al. 2004), which has provided unprecedented access to and information about uncultured microbial communities. Two studies published in 2004 demonstrated the power of this approach, particularly with simple microbial communities.
Using a whole-genome shotgun approach, Tyson et al. (2004) were able to reconstruct two almost complete genome sequences of Leptospirillum group II and Ferroplasma type II and the partial sequence of three other species from a low-complexity acid mine drainage biofilm growing underground within a pyrite ore body. This was possible because the community was dominated by a small number of distinct species. Genome analysis for each organism revealed specific pathways for carbon and nitrogen fixation and energy generation. A larger project published shortly thereafter by Venter and colleagues (2004) used a whole-genome shotgun strategy to explore the diversity in the Saargasso Sea. A total of almost 1.5 billion base pairs of DNA was generated, and it was estimated that this single sample of sea water contained ≥1800 species based on sequence relatedness. While it was not possible in this study to reconstruct nearly complete genome sequences because of the diversity of the sample, it was possible to identify >1.2 million new genes, including >700 new rhodopsin-like photoreceptors that may be involved in a new form of phototrophy in the oceans.
Because of the current limitations in assembling nearly complete genome sequence data from complex communities, there has been a renewed interest in developing methods for culturing recalcitrant microbial species (Rappe et al. 2002; Sait et al. 2002; Tyson and Banfield 2005). Several recent successes in these efforts have been reported that will most certainly facilitate follow-up functional genomics studies.
Conclusions
Although we have made tremendous progress in the past decade in the field of microbial genomics, work to date represents just the tip of the iceberg given the estimated number of microbial species on Earth. With the accumulation of more sequence data from cultivated isolates and expansion of metagenomics efforts, it is likely that the coming decade will be filled with new insights into the strange and often unpredictable microbial world. Systems-based approaches that integrate DNA sequence with data from transcriptome, proteome, and metabolome studies will begin to reveal the intricate workings of a microbial cell (Fig. 3).
Acknowledgments
I thank all of my colleagues at The Institute for Genomic Research who have contributed to TIGR's efforts in microbial genomics during the past decade, and TIGR's outside collaborators who have contributed their expertise to these projects.
Footnotes
-
E-mail cmfraser{at}tigr.org; fax (301) 838-0209.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3724205.
- Cold Spring Harbor Laboratory Press








