
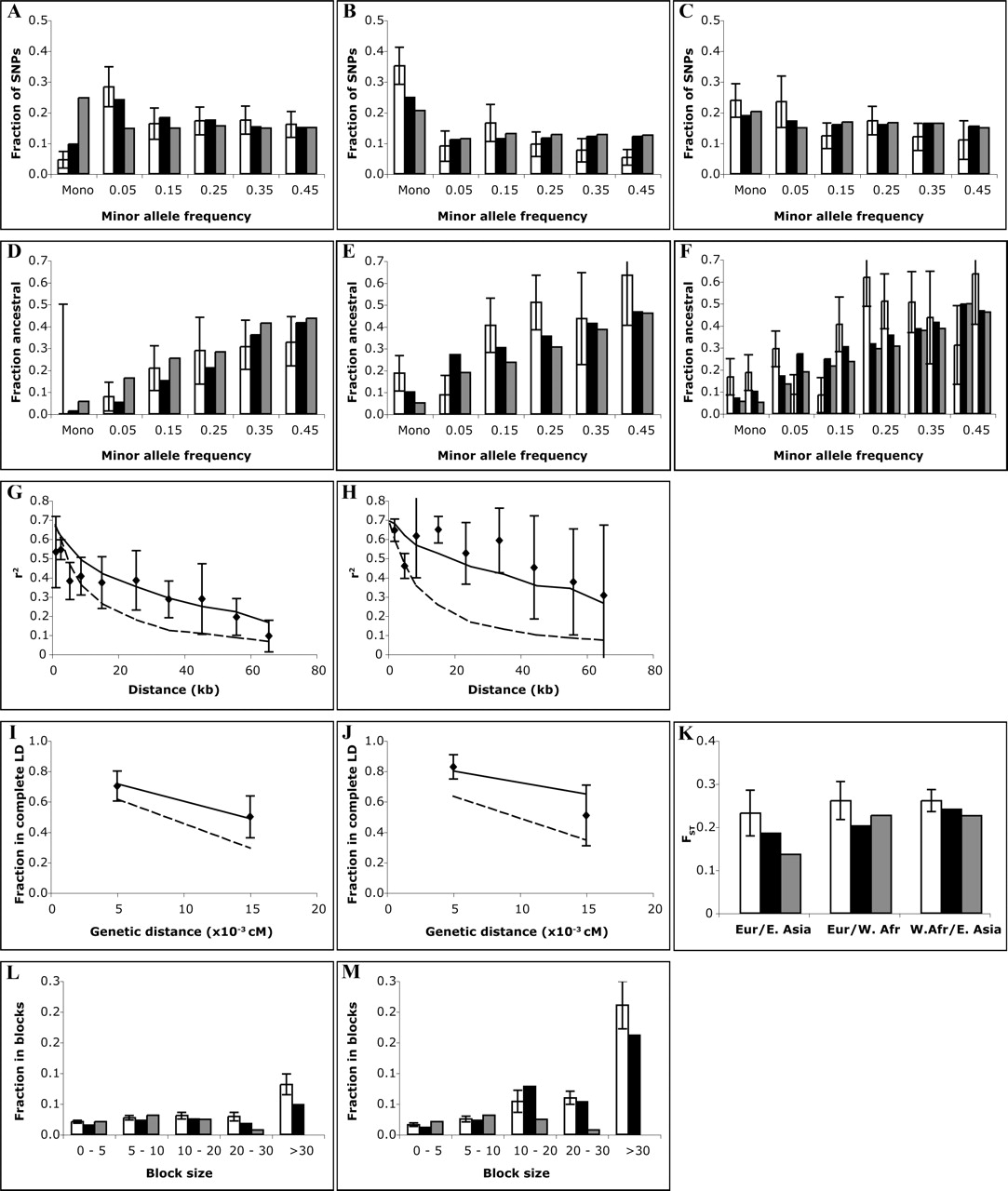
Comparison of best fit-model with empirical data, X-chromosome. Error bars represent one standard error. (A,B,C) Allele frequency spectrum. (White) Data; (black) best-fit model; (gray) standard neutral model. (A) West African. (B) East Asian. (C) European sample. (D,E,F) Fraction of alleles that are ancestral/chimpanzee, binned by allele frequency. (White) Data; (black) best-fit model; (gray) standard neutral model. (D) West African. (E) East Asian. (F) European. (G,H) Linkage disequilibrium (r2) versus physical distance. (Points) Data; (solid line) best-fit model; (dashed line) standard neutral model. (G) West African. (H) European. (East Asian omitted because of poor statistics.) (I,J) Fraction of marker pairs with perfect LD (D′ = 1.0) versus genetic distance. (I) West African. (J) European. (Points) Data; (solid line) best-fit model; (dashed line) standard neutral model. (East Asian omitted because of poor statistics.) (K) Genetic distance (FST). (White) Data; (black) best-fit model; (gray) standard neutral model. (L,M) Fraction of sequence in haplotype blocks of different sizes. (White) Data; (black) best-fit model; (gray) standard neutral model. (L) West African. (M) non-African (European + East Asian).








