
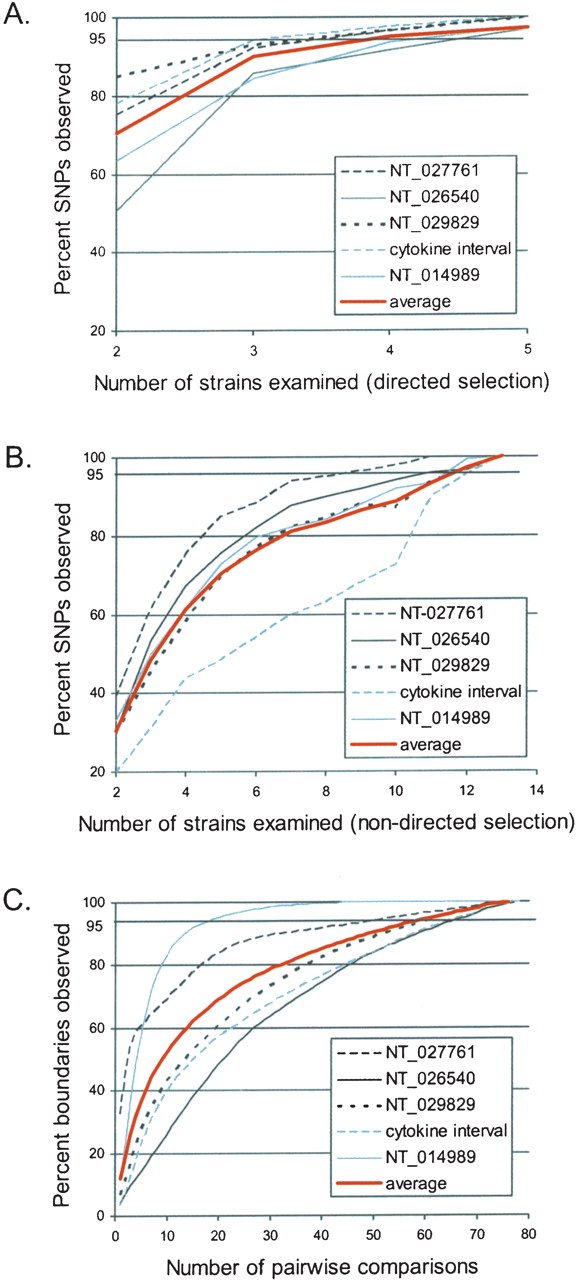
The number of strains required to capture >95% of the observed variation was calculated both by determining how many of the 13 inbred classical strains were needed to detect >95% of the SNPs (A,B) and by determining how many of the 78 pairwise comparisons of the inbred classical strains were needed to observe >95% of the segmental block boundaries (C). (A,B) The percent of SNPs observed versus the number of inbred classical strains examined was determined for each contig individually as well as the average across all five contigs. (A) Directed selection of strains based on their level of divergence (based on the Kimura 2-parameter distance), starting with the most divergent. (B) The selection of inbred strains was random (nondirected selection), with the process being repeated 100 times, and the average results for each contig and across all five contigs shown. (C) The percent of segmental boundaries observed versus the number of inbred classical pairwise comparisons examined is shown for each contig individually as well as the average across all five contigs. Intervals containing relatively few segmental blocks, such as NT_014989, require fewer pairs of strains, and intervals containing an above-average number of segmental blocks, such as NT_026540, require more pairs of strains.








