
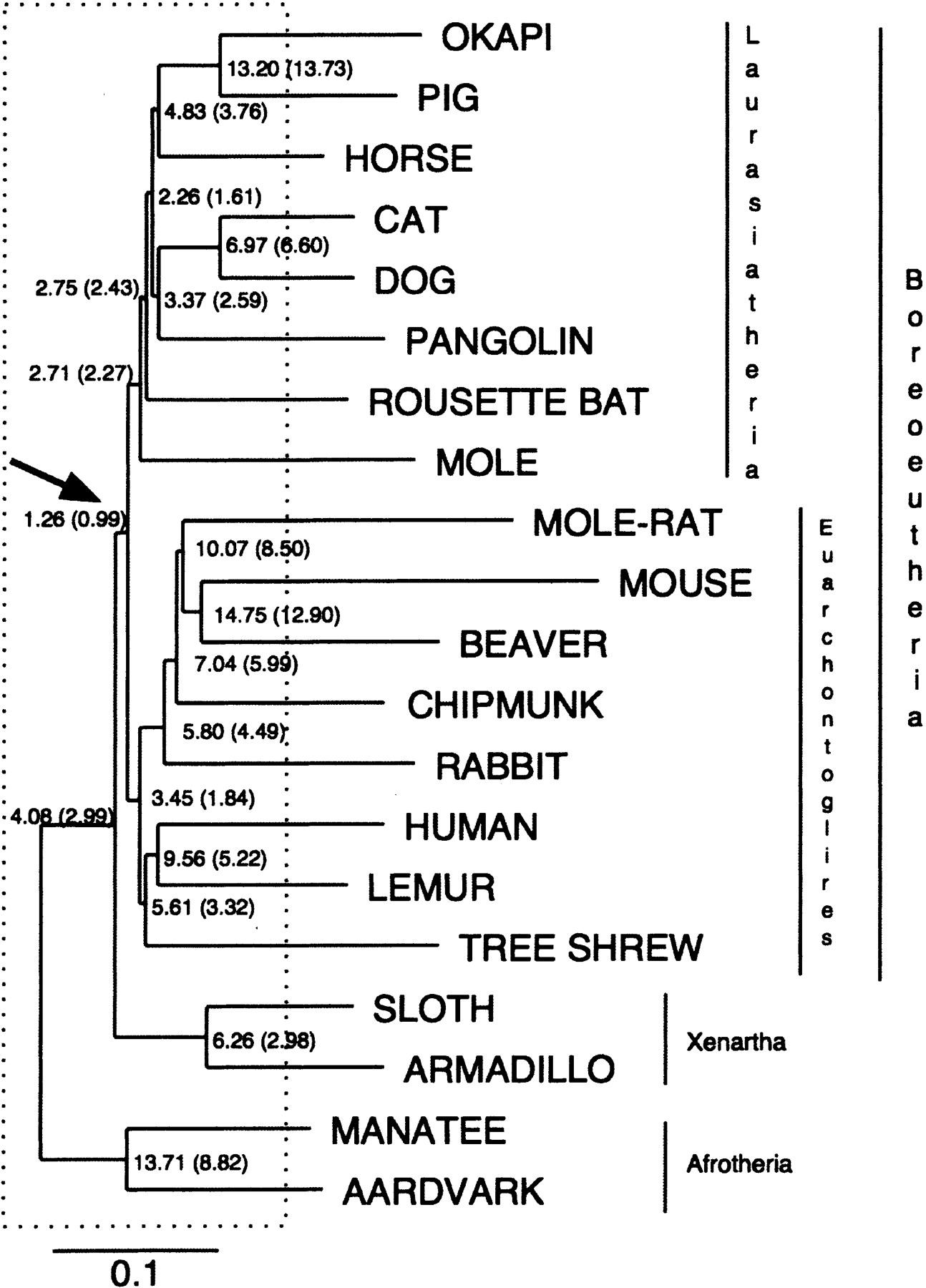
Estimated reconstructability of ancestral mammalian sequences. Average base-by-base error rate in the reconstruction of each simulated ancestral sequence. The error rate shown is the sum of the percentages of bases that are missing, added, or mismatched as a result of errors in the reconstruction, averaged over 100 simulations of sets of orthologous sequences of length ∼50 kb. Error rates are given first for all regions, and in parentheses for nonrepetitive regions only. The Boreoeutherian ancestor, which is the ancestor that can best be reconstructed, is indicated by the arrow. Branches completely located inside the box are called “early branches” (see text). The species names at the leaves only indicate what organisms we simulated; no actual biological sequences were used here. The tree topology and branch lengths are derived directly from Eizirik et al. (2001).








