
The Unusual Phylogenetic Distribution of Retrotransposons: A Hypothesis
Abstract
Retrotransposons have proliferated extensively in eukaryotic lineages; the genomes of many animals and plants comprise 50% or more retrotransposon sequences by weight. There are several persuasive arguments that the enzymatic lynchpin of retrotransposon replication, reverse transcriptase (RT), is an ancient enzyme. Moreover, the direct progenitors of retrotransposons are thought to be mobile self-splicing introns that actively propagate themselves via reverse transcription, the group II introns, also known as retrointrons. Retrointrons are represented in modern genomes in very modest numbers, and thus far, only in certain eubacterial and organellar genomes. Archaeal genomes are nearly devoid of RT in any form. In this study, I propose a model to explain this unusual distribution, and rationalize it with the proposed ancient origin of the RT gene. A cap and tail hypothesis is proposed. By this hypothesis, the specialized terminal structures of eukaryotic mRNA provide the ideal molecular environment for the lengthening, evolution, and subsequent massive expansion of highly mobile retrotransposons, leading directly to the retrotransposon-cluttered structure that typifies modern metazoan genomes and the eventual emergence of retroviruses.
The Ancient Origin of Reverse Transcriptase
There are two arguments for an ancient origin of RT. The first is theoretical and is based on the now widely accepted proposal that an RNA world preceded the form of biology with which we are familiar, the DNA world. Darnell first articulated that RT must have been present during the time of the transition between these two worlds, and therefore, must be considered ancient (Darnell and Doolittle 1986; Fig. 1). The second argument is based on the fact that RT genes are very broadly distributed among the branches of the tree of life, and have largely (but not entirely) descended vertically by descent from an ancestral RT gene (Doolittle et al. 1989; Xiong and Eickbush 1990; Eickbush 1994; Malik et al. 1999). Furthermore, the RT gene has seemingly reinvented itself in multiple and diverse forms (Boeke and Stoye 1997). In addition to the familiar retroviruses, there are pararetroviruses, which package DNA, but replicate by reverse transcription, two major classes of retrotransposons (described in the following section), as well as a more bizarre group of elements found in bacteria and organellar genomes, and hence, referred to as the prokaryotic group. The discovery of RTs in bacteria, first in the form of msDNA (short for multicopy single-strand DNA) or retron elements (Yamanaka et al. 2002) and later in the form of retrointrons (Belfort et al. 2002), provided dramatic evidence in favor of an ancient origin for RT.
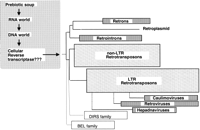
An ancient origin for reverse transcriptase. An early origin of a cellular reverse transcriptase is posited by the RNA world hypothesis (Darnell and Doolittle 1986) (left). The widespread existence of RT genes in prokaryotes, in eukaryotes, and their great molecular diversity (represented by the size of the boxes at the tip of each branch) also suggests an early origin for the RT gene. Tree diagram adapted from Eickbush (1994).
The highly diverse tree of retroelements can be rooted in the prokaryotic group of elements (Eickbush 1994). The prokaryotic group includes three types, that is, retrons, retroplasmids, and retrointrons. Retrons are RT genes that produce an unusual branched structure called msDNA made by reverse transcription of a precursor RNA primed from an internal guanosine residue— unlike other retroelements, they have no known function or ability to mobilize autonomously (Yamanaka et al. 2002). Thus far, they have been found only in a very limited subset of bacteria. Retroplasmids are known only from the mitochondria of certain fungi, replicate by reverse transcription, and exist in both circular and linear (hairpin) forms (Kuiper and Lambowitz 1988; Walther and Kennell 1999). The retrointrons, or group II introns, mobilize or retrohome to empty target sites (unspliced versions of their host genes) via a very unconventional mechanism. The excised intron lariats insert into double-stranded target DNA (copies of the DNA containing the flanking exons but lacking the intron) by reversal of the normal splicing reaction, probably aided by the maturase activity of the RT proteins encoded by these elements. They are then converted into DNA by use of a target-primed reverse transcription (TPRT) mechanism similar to that used by non-LTR retrotransposons (Zimmerly et al. 1995a,b; Yang et al. 1996). Priming is facilitated by the action of a small endonuclease domain of the RT that cleaves the intact strand of the double-stranded target DNA.
Several independent arguments strongly suggest that the prokaryotic group of RT sequences is ancestral to the RT sequences of retrotransposons and retroviruses. Counterarguments to each of these proposals exist, but as a group, these proposals are compelling. (1) It is a simple evolutionary paradigm that things evolve progressively from a simple state to an ever more complex one. Retrons, retroplasmids, and retrointrons all encode a single RT protein, often with only that enzymatic activity, whereas retrotransposons and retroviruses always encode multiple enzyme activities and usually encode multiple separate proteins. These additional activities, which include proteases, zinc finger domains, at least three distinct types of endonucleases, and integrase, appear to have been recruited from eukaryotic host genomes at multiple times in evolution, probably using the same types of mechanisms used by retroviruses when they pick up cellular oncogenes (Telesnitsky and Goff 1997). A widely accepted extension of this simple argument is that the retroviruses and pararetroviruses evolved from LTR retrotransposons by acquiring new proteins conferring the ability to efficiently leave and re-enter host cells, also known as horizontal transfer or lateral transfer (Doolittle et al. 1989). (2) The RT of one member of the prokaryotic group has the ability to perform primer-independent synthesis, similar to RNA polymerase, the presumed ancestor of RT (Wang and Lambowitz 1993). (3) The RT sequences of the prokaryotic group are the most similar to the sequences of the presumed ancestral outgroup of sequences, the RNA-directed RNA polymerases (RdRPs). Non-LTR retrotransposons, LTR retrotransposons, and retroviral RTs are progressively less closely related to RdRP sequences (Eickbush 1994). (4) The sequence of telomerase, a specialized RT considered by many to represent an ancient eukaryotic enzyme, clusters with prokaryotic and non-LTR retrotransposon RT sequences (Eickbush 1997; Nakamura and Cech 1998).
Two Types of Retrotransposons That Mobilize by Distinct Mechanisms
The retrotransposons can be divided into two major groups, the non-LTR and the LTR retrotranposons. The mechanisms of these two types of retoelements are summarized briefly here and in Figure 2. In addition, two smaller retrotransposon families, the DIRS1 (Goodwin and Poulter 2001) and BEL (Malik et al. 2000) groups, appear to be distinct, but are much less widely distributed, and thus, will not be discussed further here.
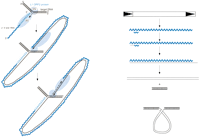
Retrotransposition mechanisms. The lifecycles of non-LTR (left) and LTR retrotransposons are outlined. Wavy lines are RNA molecules; thin black lines are cDNA strands.
Of the two major retrotransposon classes, the non-LTR retrotransposons, are less well understood mechanistically, but nevertheless, a good outline of the process exists (Kazazian and Moran 1998). The element mRNAs are translated in the cytoplasm, producing one or two proteins. One of these is a polyprotein with at least two critical activities, an endonuclease, and an RT. Most of the non-LTR elements also encode an RNA chaperone, whose role remains unclear. However, the endonuclease/RT protein is thought to bind the element RNA to form an RNP complex, which then enters the nucleus. This complex then acquires a host DNA target, in which a nick is made by the endonuclease. In a remarkable target-primed reverse transcription (TPRT) process, the 3′ OH of the cleaved target DNA primes reverse transcription of the element RNA, at or near the 3′ poly(A) end. The mechanism of the cutting of the second strand and second-strand synthesis is less well understood, but may well be symmetrical with the first, involving a second round of TPRT with the newly made DNA strand serving as template.
LTR retrotransposons move via a mechanism quite similar to that used by retroviruses. Generally, two primary protein products are made, corresponding to retroviral Gag (coat proteins) and the readthrough product Gag-Pol (RT and other enzymes). The Gag proteins together with two RNA molecules are assembled into a virus-like particle (VLP). This encapsidation may serve to further protect the element's genomic RNA molecules from degradation. Reverse transcription occurs in the VLP, and is primed by a cellular tRNA (Chapman et al. 1992) or retrotransposon RNA fragment (Levin 1995). The initial product of the RT reaction, minus strand strong-stop DNA, is transferred to the 3′ end of the RNA in a critical step that leads to subsequent completion of the minus strand DNA synthesis. If even relatively small amounts of RNA were lost exonucleolytically from the 5′ or 3′ end during this process, retrotransposition would fail. Several additional steps similar to those used by retroviruses, including a second priming event and strand transfer, lead to the final product of reverse transcription, a double-stranded DNA (Boeke and Stoye 1997; Telesnitsky and Goff 1997). RNA integrity is important for this process, which can take several hours to complete; however, a recombination-like template switching process can bypass damage to the element's RNA. The resulting DNA, together with the integrase protein (processed previously by an element-encoded protease from the RT precursor protein Gag-Pol) is transported to the nucleus, where it inserts via a transesterification reaction very similar to that used by DNA transposons (Mizuuchi and Baker 2002).
Modern-Day Distribution of RT Genes
Faced with the assumption that RT is an ancient enzyme, it becomes difficult to explain the modern-day distribution of RT genes in the three kingdoms of life, Eubacteria, Archaea, and Eukarya. The majority (67%) of sequenced eubacterial species lack a detectable RT gene in their genome (Fig. 3). For those species of eubacteria that do contain RT genes, they mostly contain only one or two RT genes. The great majority of Archaea lack recognizable RTs altogether; the only exception to this trend, Methanosarcina, has a very large genome thought to have been formed by the incorporation of a large segment of a eubacterial genome as a late lateral transfer event in its evolution (Deppenmeier et al. 2002). This species contains a set of retrointrons similar to those found in eubacteria (Dai and Zimmerly 2003). In contrast, RT genes are found in virtually all eukaryotic genomes, and are generally found in 20 to >500,000 copies per genome. Even when adjusted for genome size, eukaryotes contain significantly more RT genes. Virtually all of these are non-LTR and/or LTR retrotransposons. In a recent grand synthesis, Bushman poetically described eukaryotic genomes as “genes floating on a sea of retrotransposons” (Bushman 2002), although an astute reviewer of this work points out that genes do not float, or else gene order colinearity would not be observed in genomic comparisons. Some well-known extreme examples of this include the human genome, ∼1,000,000 non-LTR retrotransposons, SINES, and endogenous retroviruses (Smit 1996) and the maize genome, estimated to contain ∼200,000 copies of intact retrotransposons (SanMiguel et al. 1996; J. Bennetzen, pers. comm.). It is the abundance of retroelements that largely explains the C-value paradox in most metazoans. What led to such an abundance of RT genes?
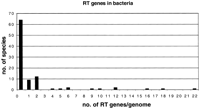
Bacterial genomes contain very few RT genes. A total of 96 completely sequenced bacterial genomes were searched by BLASTp on the comprehensive microbial resource at www.tigr.org. The two queries used were the LtrA RT from a Lactobacillus lactis group II intron (Q57005) and a retron RT from Escherichia coli (P23070). The number of BLAST hits with an E value <0.001 was tabulated for both queries, and the higher number was taken as the measure of RT gene number (visual inspection showed that this modestly inflated the number of RT genes as some of the low-scoring hits were false positives).
It could be argued that the observed discrepancy is a simple consequence of genome streamlining in bacterial genomes. Although there is no doubt that streamlining is a major evolutionary force in both eubacteria and Archaea, one can consider as a control for the above conclusion the distribution of DNA transposons among the three kingdoms. DNA transposons are found in almost all eubacterial and archaeal genomes and typically are found between 10 and 100 copies. They are also found in eukaryotes, but have a somewhat spottier distribution there, being quite well represented in certain groups (Drosophila, Caenorhabditis, Brassica), but notably absent from others (Saccharomyces, Schizosaccharomyces).
The dramatic discrepancy in retroelement distribution between prokaryotes and eukaryotes strongly suggested to me that there was some special feature(s) of being eukaryotic that represented a permissive state for RT and allowed the evolution and proliferation of retrotransposons.
The Evolution of Eukaryotes and Their Retroelements
The release of numerous eubacterial, Archaeal, and eukaryotic genome sequences has provided extensive fodder for models of how eukaryotes evolved. It is clear that we eukaryotes contain a mixture of genes descended from Archaeal and eubacterial ancestor cells (Woese et al. 1990; Margulis 1996). The precise sequence of events in the evolution of eukaryotes has been debated hotly, but a consensus is developing about the major events that must have occurred. This consensus view will be recounted here briefly.
Archaea and eubacteria were two ancient lineages of cells that had evolved distinct mechanisms of transcription and DNA replication, among other things, but otherwise shared the fundamental properties of being unicellular heterotrophs. Symbiosis of eubacterial cells (the progenitor of the mitochondrion) and Archaeal cells ultimately led to a proto-eukaryote containing a eubacterial endosymbiont. This may have begun as a casual or accidental symbiosis, but at some point, provided some important selective advantage. Several other events followed, probably involving an additional cycle(s) of acquiring additional genomes via consumption (Taylor 1974), as well as the acquisition of a number of other distinctive eukaryotic features, which will be considered separately in the next section. These events gave rise to a primitive eukaryote with the recognizable nuclear genome and mitochondrial genome, each in a membrane-bounded compartment. Acquisition of an additional photosynthetic bacterium by consumption led to a plant lineage, but for simplicity, this will not be considered further here. Because the modern eubacteria contain RT genes and the Archaea largely lack them, I will make the fairly arbitrary assumption that the same was true at the dawn of eukaryotes. The eubacterially derived endosymbiont(s) slowly transferred its genes to the nucleus of the primitive eukaryotic cell, becoming ever more dependent on its host. Remarkably, this process of gene transfer from mitochondria to nucleus is functional in modern-day yeast cells, in which the transfer of mitochondrial gene segments to the nucleus can be observed experimentally (Thorsness et al. 2002). Through this process, RT genes present as retrointrons would be transferred readily to the nucleus through this passive and stochastic process. Movement of retrointrons via homing to near-cognate sites might well have led to a proliferation of introns and the evolution of the splicing apparatus as an intron-removal mechanism. The stage was set for the evolution of retrotransposons. What specific features of eukaryotic cells made this possible?
The Nuclear Membrane
The existence of a nuclear membrane would appear to be an impediment and not a help to the evolution of retrotransposition. The translation process occurs outside of the nucleus, whereas transposition happens inside, and therefore, retrotransposons have evolved transport mechanisms to overcome these barriers. Therefore, the existence of the nuclear membrane is inhibitory to successful retrotransposition.
Linear Chromosomes
The transition to linear chromosomes from the presumed ancestral circular state may well have provided an early opportunity for an RT gene to make itself indispensable to its host by acquiring the ability to lengthen telomeres, leading to the enzyme telomerase, still the major mechanism for telomere formation in modern eukaryotes (Nakamura and Cech 1998), providing an elegant solution to the end-replication problem posed by the termini of linear DNA molecules. However, this would provide a niche for but a single copy of RT. Also, a compelling case can be made that telomerase was a relatively late acquisition, by devolution of a retrotransposon (Pardue et al. 1997), as telomeres could, in principle, solve the end-replication problem via the formation of T-loops (Griffith et al. 1999). Thus, linear chromosomes per se do not provide a compelling opportunity for the evolution of retrotransposons.
Introns
As argued above, RT may have played an important role in the widespread accumulation of introns in primitive cells, although the timing of this event has also been the subject of much debate (Gilbert and Glynias 1993; Logsdon Jr. and Palmer 1994; Stoltzfus 1994; Logsdon Jr. 1998; Simpson et al. 2002). However, the simple existence of these introns did not confer any special selective advantage on RT genes. Rather, the proliferation of introns may well be a consequence of a permissive RNA environment that allowed them to mobilize more readily in the genome.
Sex and Diploidy
Donal Hickey (Hickey 1993) and Tim Bestor (Bestor 1999) have provided eloquent arguments that the evolution of sex and diploidy provides an opportunity for mobile elements to invade host species and march inexorably to fixation in the host genome, providing they do not decrease the fitness of their host >50%. However, this argument applies to both retrotransposons and DNA transposons, and thus, is insufficient to explain the selective amplification of retrotransposons in eukaryotes.
RNA Processing Machinery
The physical separation of the processes of transcription and translation and changes in gene organization (perhaps the consequence of the nascent eukaryal nuclear genome being bombarded with fragments of its endosymbiont guest DNA), and other factors, led to important changes in the way RNA was metabolized in eukaryotic cells. The major changes were the compartmentalization of single-coding regions (by and large) into stereotypical mRNA structures punctuated by a 5′ cap structure and 3′ poly(A) tail (Fig. 4). Not only are the structures of these mRNAs prominent uniquely in eukaryotic cells, but they also coordinate to play a critical role in eukaryotic translational initiation; the 5′ cap and 3′ poly(A) interact in the cytoplasm, effectively circularizing the RNA.
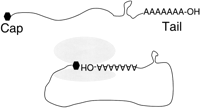
The cap and tail structure of eukaryotic mRNA.
Other Factors
There are likely to be many factors that control the proliferation of transposable elements of all types in eukaryotes. For example, organisms such as yeasts and Fugu, with smaller genomes, tend to have much higher recombination rates, and these organisms carry lighter transposon burdens. Ergo, it can be argued that such high recombination rates are inconsistent with explosive types of transposon amplification, such as has been seen in humans and maize. Very high transposon copy numbers could cause extensive secondary damage to highly recombination-proficient genomes. Similarly, diverse mechanisms controlling the copy number of certain transposons' activity, such as cosuppression of Ty1 elements in yeast (Jiang 2002) and RNAi in many eukaryotes (Ketting et al. 1999), play important roles in controlling transposon copy numbers. Whereas such factors are undoubtedly of considerable importance in determining transposon copy numbers in individual species, they do not help to explain the general trend observed that eukaryotes tend to have very high retrotransposon copy numbers relative to prokaryotes.
The cap and tail hypothesis proposes that this unique terminal structure created three special molecular opportunities for the evolution of retrotransposons. First, these termini created a very stable long-lived genomic RNA freed from the necessity to be highly folded. Second, this RNA stability facilitated the recombinational acquisition of additional host gene modules needed for the formation of retrotransposons much more likely; the long mRNAs typical of retrotransposons and retroviruses were protected from destruction by exonucleases. Third, these terminal RNA structures provided precise punctuation marks defining the retrotransposon termini and facilitating their reproduction without the loss of even a single terminal nucleotide. These traits set the stage for the evolution of elaborate and precise processes of reverse transcription evolved by retrotransposons.
Why Did Eukaryotes Evolve Caps and Tails?
A number of theories have been advanced as to the evolution of the cap and tail. Extensive work on the molecular biology of translation has shown that the 5′ cap and 3′ tail structures are directly required for initiation of translation in eukaryotes. Additionally, both RNA structures are protective against terminal degradation of the RNA. In particular, the protective role of the 5′ cap is revealed by the eukaryotic mRNA degradation pathway; this process occurs in three steps, (1) 3′ deadenylation, leading to (2) decapping, followed by (3) 5′ → 3′ exonuclease action (Tucker and Parker 2000). Although 3′ exonucleases are found in eukaryotic cells, they appear to play more specialized roles in mRNA stability, such as nonstop decay and the destabilization of specific mRNAs (van Hoof and Parker 2002).
Polyadenylation occurs in all three kingdoms of life, although it only affects a subset of mRNAs in bacteria, and actually stimulates mRNA breakdown in prokaryotes (Steege 2000). Thus, certain components of the polyadenylation machinery predated the evolution of eukaryotes, and it appears that poly(A) simply acquired new functions in eukaryotes. In the literature and in discussions with colleagues, I've become aware of three theories regarding selective pressures leading to a need for a 5′ cap. The first theory is that the compartmentalization of transcription leads to extensive opportunities for potentially inhibitory RNA folding prior to translation—potentially, such mRNA hairballs could occlude internal Shine Delgarno initiation sequences, whereas a terminal cap structure could more readily be recognized, like the end of a ball of yarn (Hershey and Merrick 2000). A second theory is that the complex nature of RNA processing in eukaryotes could lead to large numbers of misprocessed RNAs. Expression of inappropriately processed RNAs could lead to the expression of deleterious dominant negative protein fragments for example. Obviously, there are special pathways such as Nonsense-mediated decay (Frischmeyer and Dietz 1999; Gonzalez et al. 2001) and Nonstop decay, which deal with some of the RNA quality control issues raised by the existence of potentially inaccurate splicing machinery. However, a third type of proofreading is conferred by the obligatory circularization of mRNA during translational initiation—any RNA lacking intact 5′ or 3′ ends will not be translated (R. Green, pers. comm.).
Finally, Stewart Shuman has proposed that the cap arose to protect the RNA from 5′ exonuclease action, and that the latter activity represented a type of primitive immunity against RNA viruses (Shuman 2002). Thus, the Xrn1p 5′ exonuclease may have arisen in response to genomic RNA invaders, and the capping machinery evolved in parallel to protect endogenous cellular mRNAs. It is clear that eukaryotic cells evolved a series of different immunity mechanisms against invading RNA genomes, including the interferon system (Kumar and Carmichael 1998) and RNAi (Ketting et al. 1999). Needless to say, if this scenario is correct, the primitive immunity conferred by 5′ exonuclease was quickly evaded by viruses that acquired caps by various nefarious means or evolved IRES elements that bypassed the cap requirement (Shuman 2002). Nevertheless, it would appear that the acquisition of the cap/5′ exo strategy paradoxically set the stage for the evolution of a collection of internal genome invaders of eukaryotes, and eventually, retroviruses.
An interesting difference between bacteria and eukaryotes that may be related to differential RNA stability is the ability of eukaryotes to produce significantly longer proteins, such as the long polyproteins encoded by retrotransposons. Interestingly, a survey of bacterial genomes (Fig. 5) shows that bacteria, on average, encode shorter proteins than eukaryotes. This discrepancy becomes particularly acute when the longest ORFs are examined. The longest ORF in Escherichia coli K12, a putative invasin at 2383 codons, is less than half the length of the longest Saccharamyoces cerevisiae ORF, the MDN1 gene at 4910 codons, and pales in comparison to human titin at 27,118 amino acids, encoded by an astonishingly long 82-kb mRNA (Labeit and Kolmerer 1995). This limit to ORF size does not represent an absolute expression block in bacteria, as some very large ORFs encoding nonribosomal polypeptide and polyketide synthases have been discovered in various bacterial species. It is possible that the simple lifestyle of prokaryotes generally requires shorter proteins than the complex lifestyle of eukaryotes. The evolution of a more stable mRNA structure in eukaryotes may well have contributed to the evolution of much greater potential protein structure complexity in general in eukaryotes.
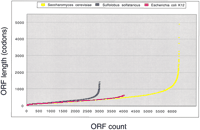
Bacteria and Archaea encode smaller proteins than eukaryotes. The number of codons in each ORF for the indicated organisms was sorted in Excel, and a point was plotted for each protein. It can be seen readily that both the mean length and total length of the eukaryotic proteins are significantly higher than those of both the eubacterial and archaeal species. Results are typical (data not shown).
Bacteria Are RNA-Hostile
Recent work on the degradation of bacterial mRNAs has elucidated the basic molecular mechanisms, which are quite different from the eukaryotic mechanism (Table 1). In summary, eubacteria like E. coli degrade their RNAs through the combined effects of multiple endonucleases and 3′ exonucleases; many of the relevant activities are organized in degradosomes (Steege 2000). One of the well-studied endonucleases, RNAse E, nicks unstructured RNA regions adjacent to structured regions. Whereas the products of such nicking are not necessarily excellent direct substrates for the 3′ exonucleases, addition of a 3′ poly(A) tail creates an opportunity to initiate the degradation process by the degradosome; hence, mRNA polyadenylation leads to degradation in eubacteria.
RNA Degradation Components Found in the Three Kingdoms of Life
If the cap and tail hypothesis is correct, it makes a number of predictions—for example, intact long RNA molecules should be difficult to detect in bacteria. It has long been known that it is extremely difficult to detect bacterial mRNAs by Northern blotting, and typical measurements of bacterial RNA half-lives range from seconds to minutes—far shorter than the half-lives of their eukaryotic counterparts, even when the mean mRNA half-life is adjusted for the cell generation time. (Fig. 6). Only a single value for average mRNA half-life is available from an Archaeal species, Sulfolobus solfataricus, which is among the slower-growing Archaea (some Archaea have fast doubling times similar to those of eubacteria), and its RNA half-life value is intermediate between eubacteria and S. cerevisiae, a eukaryote with a relatively short mRNA half-life (Bini et al. 2002). Examination of the mRNA degradation components encoded in eubacterial, Archaeal, and eukaryotic genomes shows that the eubacteria and Archaea share most of the same genes. Homologs of RNAse E, RNAse II, and polynucleotide phosphorylase are readily found by BLAST searching against Archaeal genomes. In contrast, Xrn1p homologs and capping enzyme homologs are absent from eubacteria and Archaea, but are common to all eukaryotes (Anantharaman et al. 2002). Furthermore, like eubacteria, Archaea organize at least some of their genes in operons, and use Shine-Delgarno sequences to guide ribosomes to their initiation sites, at least in some mRNAs (Shuman 2002), suggesting that translational initiation mechanisms in Archaea are more similar to those in eubacteria than in eukaryotes.
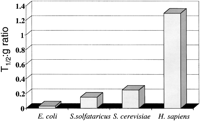
Average mRNA half lives of diverse organisms, adjusted for generation time. The data are plotted directly from Bini et al. (2002).
Eubacterial Retroelements Have Small, Highly Structured RNAs With Occluded 3′ Ends
A second prediction of the cap and tail model is that those retroelements that are found in eubacteria and Archaea will exhibit genomic features suggestive of protection against RNA degradation, such as short length, extensive secondary structure, and occluded 3′ ends. The two major classes of eubacterial retroelements display just these features. Retrointron RNA genomes are much shorter than retrotransposons and retroviruses, typically extending only 1–2 kb long versus 4–8 kb or more for typical retrotransposons and 10 kb or more for typical retroviruses. They are highly folded, their 5′ end is occluded via a 2′–5′ linkage and, moreover, they are always found in the form of a highly specific RNP, in which the RT-maturase protein is tightly bound to the intronic RNA. Importantly, the 3′ terminus of these molecules consists of a series of Watson Crick base pairs at the base of the domain VI stem of the intron, followed by two or three unpaired bases that can form a tertiary interaction with an internal segment in the intron (γ/γ′ sequences; Bonen and Vogel 2001). Similarly, the retron genome consists of a small, highly folded molecule in which the 3′ end of the RNA component is base paired to the 3′ end of the DNA component (Yamanaka et al. 2002).
Retrotransposon RNAs Are Capped and Polyadenylated
Nearly all retrotransposon RNAs contain caps and poly(A) tails, as do retroviral RNAs. The case is quite clear for LTR retrotransposons and retroviruses; there are many reports of poly(A) at the 3′ ends of LTR retrotransposon RNAs, and further evidence for posttranscriptionally added 3′ poly(A) tails in LTR retrotransposons can be found readily in EST databases. Capping is more laborious to evaluate, but some studies have been performed; for example, Ty1 mRNA was examined directly and found to be capped (Mules et al. 1998b), as are retroviral RNAs. Because LTR retrotransposons encode proteins required for their own mobility, and these must be translated from their mRNAs, it is extremely likely that all LTR-retrotransposon RNAs are capped.
One of the most important characteristics of non-LTR retrotransposons is that the vast majority of these elements actually encode poly(A) in their DNA. This 3′ poly(A) tract defines the element's 3′ end; many studies suggest that the 3′ poly(A) tract defines the site at which reverse transcription (TPRT) initiates (Moran et al. 1996). These poly(A) tails are peculiar in that they are apparently synthesized, at least in part, by RNA polymerase rather than the conventional polyadenylation machinery. However, it is possible that the 3′ poly(A) residues might be added post-transcriptionally using conventional polyadenylation. There are a few non-LTR retrotransposons such as the Drosophila I factor, which terminate not in poly(A), but in a related sequence, (TAA)n. Clearly, the 3′ end of I factor RNA is not formed by conventional polyadenylation, but by transcription. Nevertheless, the number of TAA repeats can increase during retrotransposition, suggesting that a mechanism other than conventional polyadenylation leads to the lengthening of the element 3′ end, probably slippage by the I factor RT (Pritchard et al. 1988). Interestingly, the I factor 3′ sequence can be replaced with poly(A), and the modified elements produce progeny elements with 3′ poly(A) tails (Chambeyron et al. 2002). Intriguingly, a significant subset of human L1 elements carry a related (TAAA)n repeat in place of poly(A) (Szak et al. 2002). There are a few non-LTR retrotransposons, such as the CR1 element that terminate in a 3′ terminal-repeated sequence unrelated to poly(A) (Burch et al. 1993). Presumably, these mRNAs have found another way to be circularized during translation, as they must be translated. Because this type of element lacking a poly(A)-like sequence is rare, I would propose that this is some late evolutionary adaptation. Clearly, the ancestral state of this family of elements is a 3′ poly(A)tail.
Capping, however, has not been directly studied in the non-LTR retrotransposons, although the similarity of these elements' RNAs to mRNA strongly suggests that they are capped. There is evidence that the Drosophila jockey non-LTR retrotransposon is transcribed by RNA polymerase II, which is that its mRNA synthesis is α-amanitin sensitive (Mizrokhi et al. 1988). All known pol II mRNAs are capped, therefore, non-LTR mRNAs are unlikely to be exceptions to this rule. Finally, the sequence of the human L1 element provides presumptive evidence for capping. Previous in vitro studies have shown that various RTs can readily copy the G residue comprising the cap, in spite of its unusual 5′–5′ triphosphate linkage to the mRNA (Hirzmann et al. 1993; Volloch et al. 1995; Mules et al. 1998a). The L1 sequence starts with a run of a variable number G residues, which I propose has accumulated through multiple rounds of cap reverse transcription. In support of this exotic idea, the majority of extra single nucleotides accumulated at the 5′ junction of experimentally isolated new full-length L1 insertions are G residues, whereas truncated L1 elements do not prefer single-G insertions (Symer et al. 2002; N. Gilbert, S.L. Lutz-Prigge, and J.V. Moran, pers. comm.).
A final exception to the general rule that eukaryotic retrotransposons are capped and polyadenylated is also instructive and supports the model, namely, the case of the Alu element and the related SINEs. These unusual elements don't need a cap, because they are not translated, but rely on retrotransposition proteins encoded by other non-LTR retrotransposons. Intriguingly, these elements are polyadenylated through transcription, even though they are transcribed by RNA polymerase III and, hence, are extremely unlikely to interact with the polyadenylation apparatus. However, these pol III transcripts lack a 5′ cap. A different mechanism of protection from 5′ exonuclease is adopted by these elements; as in the case of eubacterial retroelement 3′ ends, Alu and related retroelements, as well as the tRNA-derived retroelements, are also highly folded and the 5′ end of the RNA is always found in an extensively base-paired structure (e.g., see Sinnett et al 1992), which would protect it against Xrn1-like 5′ exonucleases.
Retrotransposon RNA levels are highly variable and tend to be tissue specific in metazoans, with high levels reached only in the germ line in most cases (Chaboissier et al. 1990; Branciforte and Martin 1994). Naturally, the abundance of retrotransposon RNAs is very strongly correlated with retrotransposition frequency. Because retrotransposition frequencies are set by some complex evolutionary interplay unique to each host/retrotransposon combination, it is not surprising that there is great variability in retrotransposon RNA levels. Nevertheless, there are some very dramatic cases of very high retrotransposon RNA levels that provide strong evidence that the cap and tail structure are compatible with high levels of retrotransposon transcript stability. Of note, the Drosophila retrotransposon copia is so named because of its incredibly copious mRNA (Young and Hogness 1977), and yeast Ty1 mRNA levels are among the most abundant in the yeast cell (Curcio et al. 1990), with Ty1 mRNA visible as a discrete band in poly(A)-selected RNA preparations.
In conclusion, the stable and well-punctuated mRNA system was probably critical in allowing eukaryotes to evolve an ever more complex lifestyle, permitting longer more complex proteins and increased molecular diversity through alternative splicing. This same key change probably led to the extensive proliferation of retroelements, including retroviruses, in the many complex guises in which they are found today.
Acknowledgments
I thank Laurel Ricucci for help with the figures, and members of my laboratory for helpful discussions. Research was supported with grants from the NIH.
Footnotes
-
E-MAIL jboeke{at}jhmi.edu; FAX (410) 614-2987.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1392003.
- Cold Spring Harbor Laboratory Press








