
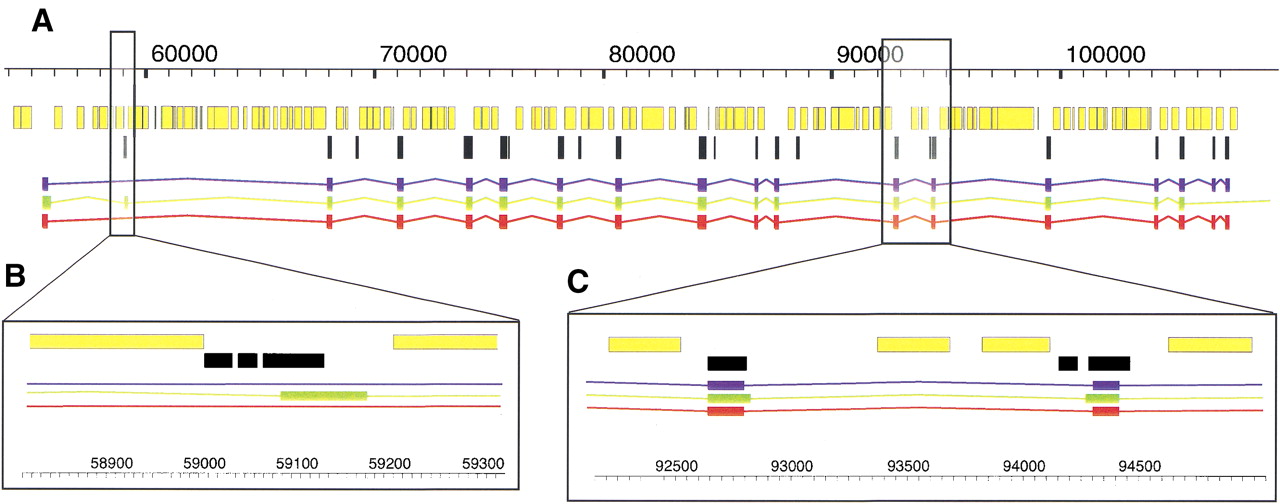
A detailed view of a TWINSCAN prediction (red), a GENSCAN prediction (green), and an aligned RefSeq transcript (blue). Masked repetitive and low-complexity regions (yellow) and mouse alignments (black) are indicated. (A) Complete gene prediction at theKIAA1630 gene (NM_018706) from Homo sapiens 10p14. Note that the presence of conservation is neither a necessary (e.g., the first exon), nor a sufficient (e.g., the first alignment block condition for TWINSCAN to predict an exon. (B) A magnified region around the second exon predicted by GENSCAN. TWINSCAN correctly omits this exon because the conserved region ends within it. (C) A magnified region around the 11th and 12th RefSeq exons. TWINSCAN correctly predicts both splice sites because they are within the aligned regions. These images were produced with AceDB (http://www.acedb.org/).








