
Intraproteomic Networks: New Forays Into Predicting Interaction Partners
Biological treasure troves of complete protein complements of diverse organisms (proteomes) have been unveiled in the past few years as a result of the tremendous success of genome projects. The fundamental fascination of most biochemists and molecular biologists is how the different polypeptides comprising the proteome interact to conduct “business” in various biological systems. The flood of genomic data has made large-scale attacks on this problem through computational and experimental methods very feasible. On the computational side, the main progress has been in the form of identification and classification of the individual protein domains, thereby helping to narrow down to the actual determinants of the intraproteomic interactions (Ponting et al. 2000; Lander et al. 2001). On the experimental side, high-throughput proteomic analysis has yielded protein-interaction maps for different organisms at an unprecedented level of detail (Matthews et al. 2001; Tucker et al. 2001). Initial analysis of this data reveals that the interactions within the proteome of an organism constitute a scale-free network characterized by hubs of highly connected polypeptides, each of which interact with several proteins with few or no further connections (Snel et al. 2002; Wolf et al. 2002).
Despite these advances, the precise set of changing interactions that are related to the organism's responses to changing environments, or those that are involved in development and differentiation of multicellular organisms, is not easily deduced from these studies. Furthermore, the exact determinants of the interactions in a polypeptide and the effects of modifications on them cannot be extrapolated directly from these large-scale studies. This is where a new genre of computational studies could provide potentially interesting results. Essentially, these studies would need to go beyond the identification of the individual modules involved in interactions and predict some of the actual interactions themselves. While the great structural diversity of the protein domains makes this task rather enormous, the current availability of large amounts of structural data makes this, in part, tractable. Computational analysis of this problem also is likely to uncover several general principles behind protein interactions that are unlikely to be directly uncovered through other methods.
Proteins mediate interactions with each other and other molecules via a great diversity of interfaces that span the whole range of structural complexity from simple α-helical surfaces, through repetitive α-helical or β-propeller superstructures, to complex binding pockets. One of the simplest interaction interfaces seen in proteins is the coiled coil that comprises two α-helical stretches winding around each other to form a double-helical superstructure (Fig.1) (Lupas 1996; Burkhard et al. 2001). The coiled-coil regions are characterized by heptad periodicity and typically contain hydrophobic residues (like leucine) that lie on the same side of the helix and stabilize the superstructure through hydrophobic interactions (Fig.1). As a result, these structures often are referred to as leucine zippers (O'Shea et al. 1989) and are utilized extensively in homo- or heterodimerization or oligomerization in all life forms, especially in eukaryotes. A number of eukaryotic transcription factors combine a DNA-binding module, such as basic stretch (B-ZIP) (Landschulz et al. 1988), basic helix-loop-helix domain (bHLH) (Blackwell et al. 1990), or a homeodomain (HD-ZIP) (Schena and Davis 1992), with a coiled-coil region. The simplicity of the interaction interface and availability of extensive biochemical studies on the dimerization of the B-ZIP transcription factors make them attractive targets for prediction of protein-protein interactions through computational analysis of their sequence and structure.

A representation of interactions in a coiled coil. This coiled coil is the basic stretch (B-ZIP) module of the transcription factor Pap1 and shows three different kinds of stabilizing interactions between residues of the heptad of each interacting partner. The leucines of the leucine zipper in position four of the heptad are shown in yellow, an example of the attractive interaction between the residues of the fifth and seventh position is shown in red, and the interaction between the residue pair in the first position of the heptad is shown in violet.
Fassler et al. (2002) present results in this direction by using biochemical studies and thermodynamic measurements to identify two simple principles that govern B-ZIP dimerization. They suggest: (i) The presence of oppositely charged residues on the respective fifth and the seventh positions of the two intertwining heptads result in an attractive interaction favoring formation of a dimeric pair, while residues with the same charge in these positions result in a repulsion that acts against their dimerization (Fig. 1). (ii) Residues in the first position of the heptad of one monomer interact with the corresponding residues in the first position in the second monomer (Fig. 1). Polar or aliphatic residues in these positions stabilize dimers to a greater extent when they interact with the same kind of residue, as against pairs that may have an aliphatic-polar residue interface. Putting these principles together, Fassler et al. (2002)present extrapolations for the dimerization specificities of B-ZIP proteins from the complete proteome of Drosophila melanogaster. The authors observe that a large number of theDrosophila B-ZIP proteins contain a polar residue (usually asparagines) in the first position of their heptad repeat. Combining this with the charge states in the fifth and seventh positions of the heptads they suggest that many of these proteins are more likely to homodimerize rather than heterodimerize with other B-ZIP proteins.Jra and Kay, the Drosophila orthologs of human protooncogene products, Jun and Fos, are predicted to heterodimerize rather than homodimerize based on the presence of repulsive residues in the fifth and seventh positions. Consistent with their model, these Drosophila proteins as well as their human orthologs have been experimentally shown to dimerize.
A large number of B-ZIP, bHLH-ZIP, and HD-ZIP transcription factors are encoded by most of the crown-group eukaryotes, especially the plants and humans (Riechmann et al. 2000; Lander et al. 2001), and the majority of them remain uncharacterized in terms of their interaction partners. Thus, extensions of studies such as those presented byFassler et al. (2002) might aid in uncovering the diversity of their interactions and also understanding how these interactions have changed over evolution. Additionally, in eukaryotes, coiled coils act as interfaces for dimerization in proteins such as the cytoskeletal intermediate filaments; motor proteins like myosin, kinesin, and dynein; the membrane fusion proteins like SNAREs; chromosome condensation proteins like SMC; and the secretory vesicle cargo packaging proteins like P24 (Burkhard et al. 2001). Further analyses on the lines of those carried out on the B-ZIP transcription factors also may be useful in unraveling the range of interactions between these major functional components of the cells.
Can analogous simple rules be of value in predicting interactions between more complex protein interfaces? Preliminary results suggest that rules, with some degree of discrimination, may be devised for slightly more complex interaction modules, for which some biochemical data exist. One such module is the MYB-like domain that contains a version of the Helix-turn-Helix (HTH) fold. These modules have been known to interact with either DNA or proteins. They interact with DNA by inserting the “recognition helix” of the HTH into the major groove of the DNA and some forms additionally interact with the minor groove via basic residues from the N-terminal tail (Hanaoka et al. 2001). Based on these properties, it has been proposed that the DNA-binding versions of the MYB domain form a strong basic surface on the side that interacts with DNA, while those that do not bind DNA (often referred to as SANT domains [Aasland et al. 1996]) instead, have a corresponding acidic or mixed charged surface (Hanaoka et al. 2001). Thus one could use models of MYB domains showing the surface electrostatic potential and overall positive charge in the domain as potential predictors for their interactions. Comparison of these properties between classic DNA-binding versions of the MYB domain and the SANT domains, which are found in numerous chromosomal proteins, indicates that many of the latter contain strongly acidic surfaces in place of the basic DNA-binding surfaces of the former (Fig.2A). Consistent with this, SANT domains of proteins such as ADA2p and TFIIB“ interact with proteins rather than DNA (Shah et al. 1999; Sterner et al. 2002). The overall acidic surface (Fig. 2) predicts that these are likely to interact with basic targets on the partner proteins. Further biochemical investigations of these domains may help in obtaining more specific rules for their interactions.
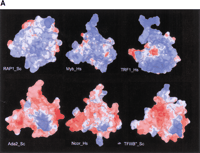

(A) Distribution of surface electrostatic potential on the DNA binding interface of Myb/SANT domains. The top row shows DNA binding Myb domains that have a basic surface charge (blue) on their DNA binding interface, and the bottom row shows SANT domains known to be involved in protein-protein interactions that have an acidic (red) or mixed charged surface in the same region. (B) Multiple sequence alignment of a representative set of DNA binding Myb domains (top) and SANT domains (bottom). Proteins are denoted by their gene names or pdb id (where a structure is available), their species abbreviations, and the Genbank Identifier (gi). The coloring reflects the amino-acid conservation profile at 90% consensus. The + charge in the minor groove binding site is shown to the right. Species abbreviations are as follows: At, Arabidopsis thaliana; Bs,Bacillus subtilis; Dm, Drosophila melanogaster; Gaga,Gallus gallus; Hs, Homo sapiens; Mm, Mus musculus; Sc, Saccharomyces cerevisiae; Zm, Zea mays.
Thus, there is some promise that, at least some of the interactions mediated by compact domains with well-characterized structures, such as the HTH, the bHLH domain, the Bromodomain, or the Chromodomain, also could be captured through relatively simple rules. However, this would depend heavily on robust experimental evaluation of specific interactions to provide sufficient precedence to develop useful rules. While this experimental aspect is not particularly advanced for a large number of the characterized domains, future opportunities in this direction could emerge from the collusion of sequence and structure studies of specific protein domains with the protein-interaction maps generated by high-throughput proteomics.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL aravind{at}ncbi.nlm.nih.gov; FAX (301) 480-9241.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.353302.
- Cold Spring Harbor Laboratory Press








