
New Twists on the Epigenetics of CpG Islands
CpG islands are >200-bp stretches of DNA that have a significantly higher concentration of CpG dinucleotides than the bulk of the genome. Whereas 70%–80% of all CpG dinucleotides in the human genome are methylated, CpG islands by and large remain unmethylated (Cross and Bird 1995), with the exception of those associated with imprinted and X-linked genes (Razin and Cedar 1994). In this issue ofGenome Research, Strichman-Almashanu et al. (2002) present the first successful systematic approach to generating libraries of differentially methylated and unique CpG islands and show its use in uncovering novel imprinted genes.
Methylated CpG Islands and Genomic Imprinting
Figure 1 outlines the strategy used to select fragments that were subsequently cloned and sequenced. Whereas the majority of the clones isolated in this manner corresponded to CpG islands, most of the several hundred clones represented CpG island repeats, such as the nontranscribed intergenic spacer of ribosomal DNA and a transposon repeat, termed SVA. The library was further processed, therefore, to generate clones that were all determined to be CpG islands. Southern analysis of the library revealed that the clones fell into two categories: those that are densely methylated on both alleles both in soma and in sperm (termed SMRs) and those differentially methylated (termed gDMRs), as determined by analyzing DNA of uniparental tissues presumably displaying epigenetic states of maternal or paternal origin.
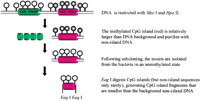
Generation of differentially methylated CpG island library. Unfilled lollipops represent unmethylated CpGs and filled lollipops represent methylated CpGs. See text for details. (Revised from Fig. 1 ofStrichman-Almashanu et al. 2002, with permission.)
One of the differentially methylated sequences of the CpG island library of Strichman-Almashanu et al. (2002) mapped to a previously known imprinted gene, HYMA1. Encouraged by this finding, the authors searched their library for neighboring genes that were expressed in a parent of origin-dependent manner. Indeed, a novel imprinted gene, termed Elongin A3, was found flanking another differentially methylated CpG island sequence of the library. This confirmed not only the supposition that differentially methylated CpG islands might be common among imprinted loci but also the feasibility of the approach of Strichman-Almashanu et al. to identify novel imprinted genes.
Genome Structure and Epigenetic Status of CpG Islands
It has recently been proposed that CpG islands fall into several groups, one of which represents unique CpG and generally unmethylated islands associated with the 5′ region of housekeeping genes, whereas another includes high-copy nongene CpG islands that are dominated byAlu I repeat elements (Ponger et al. 2001). BecauseAlu I repeats are generally methylated and transcriptionally silent, high-copy CpG islands are predicted to be methylated. Indeed, the report of Strichman-Almashanu et al. (2002) identified one of the high-copy CpG islands (SVA) to be heavily methylated. This observation is not surprising given that repeat sequences provide signatures for de novo methylation, according to the host defense model (Bestor and Tycko 1996).
Strichman-Almashanu et al. (2002) also report the existence of a new class of unique CpG islands that are methylated on both alleles in all tissues examined. Interestingly, these CpG islands (SMRs) mapped to isochores with high GC content (>0.5), whereas the differentially methylated islands (gDMRs) were concentrated in isochores with low GC content (<0.5). The class of unmethylated or differentially methylated CpG islands could stand out in a CpG-less environment and provide landmarks for various recognition events, such as the initiation of chromatin condensation by TP2 during spermiogenesis (Kundu and Rao 1996). The complexity of CpG island compartmentalization of the mammalian genome was further emphasized by the observation that the methylated high-copy CpG islands frequently localize close to telomeric ends (Strichman-Almanshanu et al. 2002), as do densely methylated nonisland CpG stretches (Brock et al. 1999), indicating some methylation-dependent role in chromosomal integrity. This deduction is supported by the observation that DNA methyltransferase, Dnmt1, is essential for genomic stability in mouse embryonic stem cells (Chen et al. 1998).
Differentially Methylated CpG Islands and Expression Domains
Perhaps the most intriguing functional aspect of the epigenetic states of CpG islands lies in their control of expression domains, as exemplified by the close apposition of differentially methylated CpG islands to imprinted genes and the distribution of unique and unmethylated CpG islands to the promoters of 50%–60% of all human genes (Cross and Bird 1995). Taken together, the data indicate that protection against de novo methylation is likely to have involved functional selection based on gene expression patterns. Whereas it is widely argued that unmethylated CpG islands stably propagate an open chromatin conformation that allows trans-acting factors to access pivotal promoter elements, this might be an oversimplification, as hinted at by Strichman-Almashanu et al. (2002).
Under the host-defense model, which posits that high-copy number repeats provide signals for de novo methyltransferases, methylation patterns should spread from high-copy repeats scattered throughout the genome (Bestor and Tycko 1996). The resistance of many of the unique CpG islands to waves of de novo methylation is reminiscent of how chromatin insulators are perceived to prevent silencers from accessing gene promoters. Indeed, unpublished observations (Strichman-Almanshanu et al. 2002) indicate that many CpG islands of the Strichman-Almanshanu et al. report interact with the 11-zinc finger protein CTCF, which organizes chromatin insulator functions (Ohlsson et al. 2001). An alternative view of the role of unmethylated or differentially methylated CpG islands invokes, therefore, protection against silencers and/or enhancers in a gene context-dependent and stably heritable manner, as a contribution to transcriptional regulation (Fig. 2) (Hejnar et al. 2001). Interestingly, CTCF is known to participate in the generation of the methylation-free domain of the maternally inherited imprinting control region flanking the H19 gene (V. Pant et al., unpublished observations) and, hence, might also explain the methylation privilege status of such CpG islands. At the same time it should be noted that only a fraction of CpG islands were found to interact with CTCF in an array-based approach (Mukhopadhyay et al. unpublished observation), indicating that several additional factors functionally interact with discrete subclasses of CpG islands. Likely candidates include the Sp1 family of factors interacting with CpG-rich sequences (Brandeis et al. 1994; Macleod et al. 1994) and other CpG-binding factors, such as hCGBP (Voo et al. 2000).

De novo methylation of CpG islands during cancer might silence neighbouring genes in two distinct manners.
Cancer and Epigenetic States of CpG Islands
Since the initial discovery of epigenetic lesions in cancer cells by Feinberg and Vogelstein (1983), it is now abundantly clear that epigenetic marks can be very unstable in cancer (Issa and Baylin 1996), creating patterns of epigenetic mosaicism (Ohlsson et al. 1999). The normally unmethylated CpG islands, for example, are often methylated in cancer to silence flanking genes during neoplasia (Fig. 2). The existence of a class of normally methylated CpG islands as reported byStrichman-Almashanu et al. (2002) indicates the complementary scenario that demethylation of CpG islands can lead to unscheduled activation of genes. This was first shown at the MAGE-1 locus, which is normally expressed only in the male germline but is activated in human tumors (De Smet et al. 1996). However, as pointed out byStrichman-Almashanu et al. (2002), these scenarios are not straightforward because their normally methylated CpG islands remained methylated in cancer cells, that is, Wilms' tumors. Indeed, it has been reported that the patterns of cancer-associated methylation of CpG islands depend on several factors, such as cell lineage and environmental stimuli (Yan et al. 2001). The most plausible explanation for these observations posits the existence of a repertoire of factors that govern the epigenetic status of different subsets of CpG islands. The genetic or epigenetic inactivation of such factor functions during neoplasia might trigger epigenetic lesions, therefore, which are specific for each group of CpG islands. This in turn might expose the genes flanking such islands to silencers (or enhancers) and set the stage for various selection pathways.
OUTLOOK
Thanks to the work by Strichman-Almashanu et al. and others, a fine-scale structure of CpG islands is emerging, with classes of islands characterized according to genomic location and epigenetic status. However, although classifying CpG islands on the basis of epigenetic status in the germline and soma looks appealing, we must be aware that methylation status is a dynamic feature that is reshaped at several different stages during development and in cancer. The function of CpG islands during these events and the contribution of their epigenetic states to the structure and function of the genome will be sure to provide fresh insight into the mechanisms of development and disease.
Footnotes
-
↵1 Corresponding author.
-
E-Mail: Rolf.Ohlsson{at}ebc.uu.se; FAX 46 18 4712683
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.18002.
- Cold Spring Harbor Laboratory Press








