
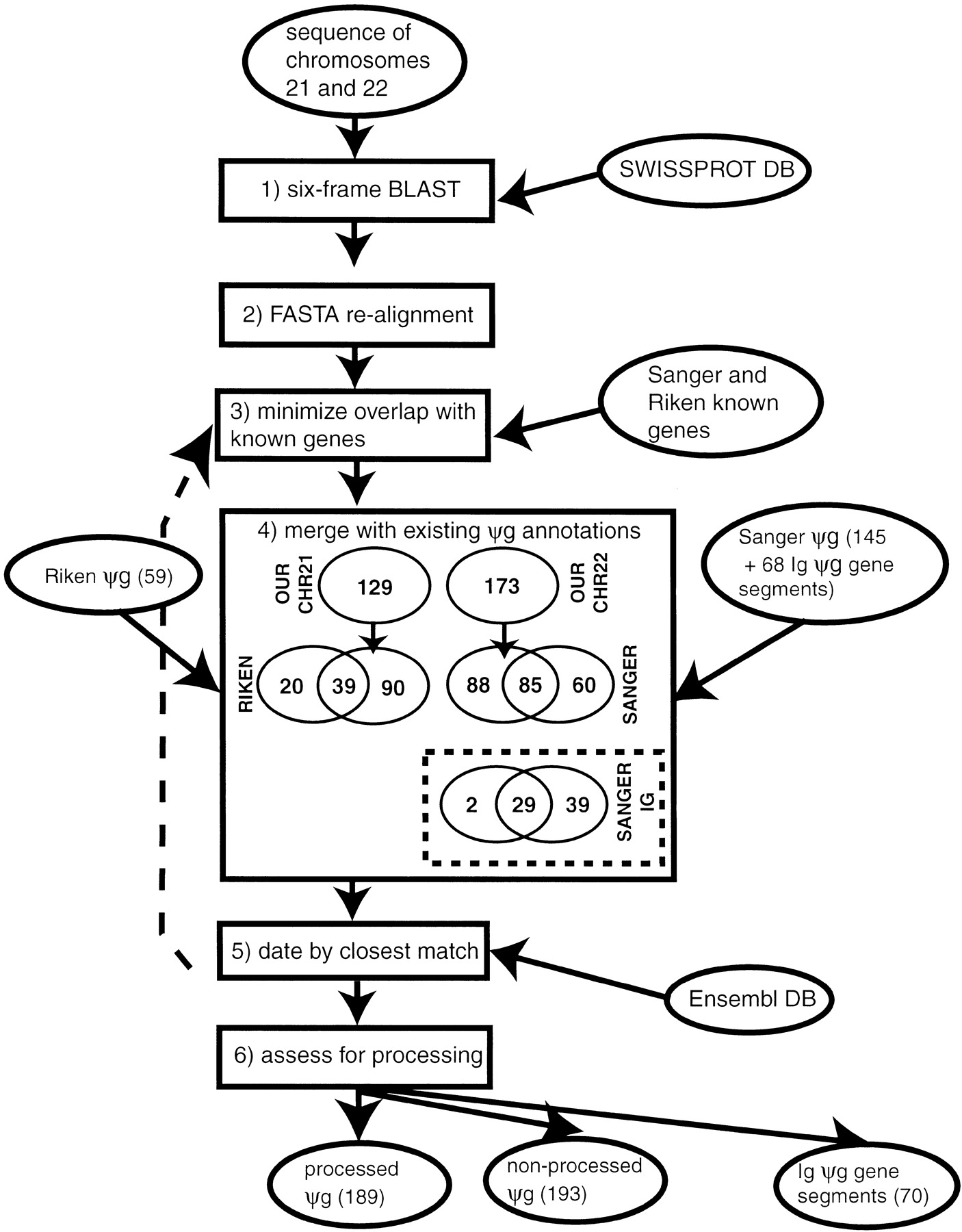
Flow diagram of the scheme for assignment of pseudogenes. The schematic shows the steps in assignment of pseudogenes. Ovals denote sources of data, and boxes denote operations. The term “ψg” denotes “pseudogene.” The steps are as follows (described in detail in the text): (1) Six-frame blast. Comparison of SWISSPROT database to chromosome 21 and 22 genomic sequences using BLAST(Altschul et al. 1997) to find potential pseudogenic protein homologies (with stop codons in them). (2) FASTA realignment. Realignment with the FASTA package (Pearson et al. 1997) of the top-matching sequence for the potential pseudogene to find longest protein-homology fragment that has >1 disablement (frameshift or premature stop codon). (3) Minimize overlap with known genes. Overlap of putative pseudogenes with known human genes (from the Sanger center annotations for chromosome 22 and the Riken center annotations for 21) is minimized by choosing a suitable margin at the ends of pseudogenic homologies within which to ignore disablements. (4) Merge with existing ψg annotations. Pseudogene annotations from the Sanger and Riken centers are merged with those that are duplicates of these in our own set of annotations being deleted. (5) Date by finding closest matching Ensembl protein. For each pseudogene, the closest matching Ensembl human protein was found so that the pseudogene could be approximately dated. This was realigned to the genomic DNA sequence (backward step denoted by dotted arrow) and used as a replacement if it produced a longer pseudogene. (6) Assess for processing. All pseudogenes were then assessed for processing by searching for evidence of polyadenylation or extensive spans of protein homology in the absence of exon structure.








