
Pixelating the Brain
This layer presents a great variety of forms and of sizes of cells. In general, the cells of which it is composed are more or less globular and much inferior in size to the large pyramid. But it is also possible to see, as did Golgi, who has recognized them before in man and higher mammals, a great number of fusiform cells and also true pyramids oriented in the same manner as the cells of the second and third layers.
—Santiago Ramon y Cajal (describing neuronal cell types in cortical layers 2 and 3)
Gene expression profiling provides a powerful tool for examining alterations in the state of a cell. First in yeast, and increasingly in more complex systems, expression profiling is providing a unique means of rapidly identifying specific genes whose expression is altered (Brown and Botstein 1999; Sorlie et al. 2001). Perhaps more importantly, it provides the means of gaining a broad perspective on changes that involve networks made up of many genes (Kim et al. 2001).
The central nervous system provides a unique set of challenges for the application of expression profiling technologies. First and foremost is the overwhelming number of cell types that are present in the brain. Each cell type makes up only a small fraction of the total, making it impossible to identify minority opinions with a simple polling strategy. The most common method of dealing with this problem is to survey local opinion, by dissecting out a few select structures, with the idea that this will allow minority voices to rise above the background roar. It is not always possible to know which are the relevant brain regions and in addition, the importance of standardization in tissue sampling protocols has been ignored.
Brown et al. (2002), in this issue, have taken a brave stab at solving these problems with a method they call ‘voxelation.’ Applied to human brain sections, voxelation divides a thick brain section into squares using a scalable grid. RNA harvested from the resulting cubes of tissue (voxels) is used for expression profiling. This allows the same regions to be compared between two different brains in a standardized fashion and at the same time allows an unbiased sampling of many regions at once (Fig. 1). In the present case, the authors examined the pattern of gene expression in an individual plane of human brains. Tissue was harvested from both normal and Alzheimer's diseased brains, and two types of comparisons were made. Different brain regions were compared to each other, and specific brain regions were compared between normal and diseased brains. It is to be expected that there are differences in gene expression between different brain regions and between normal and diseased brain. The clear differences that emerged provide a pleasing validation of the methodology. The implication of these differences for the different function of brain regions and for the loss of function in the Alzheimer's brain will require further study.
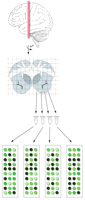
Voxelation is a technique for analyzing gene expression across large brain regions in a standardized fashion. Thick brain slices are dissected using a scalable grid. RNA prepared from each piece of brain tissue is used in micro-array hybridization experiments.
More important than the specific differences that were found is the suggestion of what might be accomplished in the future. A careful application of the method to sections covering the entire brain promises to allow three-dimensional atlases of gene expression to be constructed, similar to the activity maps made using functional magnetic resonance imaging or positron emission tomography (whence comes the term voxel). The present investigation studied a set of about 5000 genes and did not use the most sensitive means of producing probes from the harvested tissue. Conceptually straightforward extensions of the technique would involve sampling a transcript from each of the approximately 30,000 genes. Available methods for amplifying the probe RNA would allow the amount of tissue to be reduced to the point that 325,000 voxels from an adult human brain could be sampled. An atlas of gene expression produced by this approach would be an enormous data set of 10 billion data points. An experiment of this size is not one that we can imagine will be done routinely in the near future, and the cost per voxel would have to drop to the $1–$10 range to be considered. Nonetheless, a dense voxel sampling of several normal brains would provide a useful set of reference data. Sampling of gene expression in small sets of standardized voxels in diseased brains would then allow comparison to the standard data and to other experiments. This would provide an enormous boost to our ability to analyze alterations in gene expression in disease states.
There are still further technical innovations that need to occur, though, to allow the full power of expression atlases to be realized. The most important disease model that we have is the laboratory mouse. Comparisons of patterns of gene expression between diseased human brain and diseased model brain will require a corresponding standard voxel set and atlas of the mouse brain. Here our furry friend provides a problem with his small size and correspondingly small brain. Each structure in the human brain is miniaturized in the mouse brain at a scale of about 1:55. This means that an atlas of the mouse brain that would be in register with the 325,000 voxel human atlas imagined above would require amplification techniques above and beyond those applied to the human. More likely, it seems, would be that the information from the human atlas would flow down and inform the design of experiments in the mouse that look at specific regions and sets of genes. Quantitative PCR machines are available that can look at the expression of 96 genes in parallel. Small sets of genes looked at in this fashion could allow direct comparisons between disease and disease model.
Parallel atlases of mouse and human would be tremendously valuable for another reason as well. The mouse provides the opportunity to identify the specific cell types within an anatomical structure that are providing the signal—the opportunity, in other words, to let the minority voice sing out in solo. Increasingly, Cre lines are being used to label specific cell populations (Nagy 2000). If the techniques are adapted appropriately, it should be possible to purify these labeled cells. The application of expression profiling to these cells would then finally break down the background roar into individual voices. By reference to expression atlases, this would allow the mouse to predict specific cells in the diseased brain that are affected and would take us far down the road to the molecular dissection of diseases affecting our most complex organ—the brain.
Footnotes
-
E-MAIL andpete{at}itsa.ucsf.edu; FAX (510) 985-3101.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.227802.
-
E-MAIL andpete{at}itsa.ucsf.edu; FAX (510) 985-3101.
-
Article and publication are athttp://www.genome.org/cgidoi/10.1101/gr.227802.
- Cold Spring Harbor Laboratory Press








