
A Marker-Dense Physical Map of the Bradyrhizobium japonicum Genome
- Jeffrey P. Tomkins1,4,
- Todd C. Wood1,
- Minviluz G. Stacey2,
- John T. Loh2,
- Adam Judd3,
- Jose L. Goicoechea1,
- Gary Stacey2,
- Michael J. Sadowsky3, and
- Rod A. Wing1
- 1Clemson University Genomics Institute, Clemson, South Carolina 29634, USA; 2Center for Legume Research, Department of Microbiology, University of Tennessee, Knoxville, Tennessee 37996, USA; 3Department of Soil, Water and Climate, Biological Process Institute and Center for Microbial and Plant Genomics, University of Minnesota, St. Paul, Minnesota 55113, USA
Abstract
Bacterial artificial chromosome (BAC) clones are effective mapping and sequencing reagents for use with a wide variety of small and large genomes. This report describes the development of a physical framework for the genome of Bradyrhizobium japonicum, the nitrogen-fixing symbiont of soybean. A BAC library for B. japonicum was constructed that provides a 77-fold genome coverage based on an estimated genome size of 8.7 Mb. The library contains 4608 clones with an average insert size of 146 kb. To generate a physical map, the entire library was fingerprinted with HindIII, and the fingerprinted clones were assembled into contigs using theFingerprint Contig software (FPC; Sanger Centre, UK). The FPC analysis placed 3410 clones in six large contigs. The ends of 1152 BAC inserts were sequenced to generate a sequence-tagged connector (STC) framework. To join and orient the contigs, high-density BAC colony filters were probed with 41 known gene probes and 17 end sequences from contig boundaries. STC sequences were searched against the public databases using FASTA andBLASTX algorithms. Query results allowed the identification of 113 high probability matches with putative functional identities that were placed on the physical map. Combined with the hybridization data, a high-resolution physical map with 194 positioned markers represented in two large contigs was developed, providing a marker every 45 kb. Of these markers, 177 are known or putative B. japonicum genes. Additionally, 1338 significant BLASTXresults (E < 10−4) were manually sorted by function to produce a functionally categorized database of relevantB. japonicum STC sequences that can also be traced to specific locations in the physical map.
The availability of microbial genome sequences is revolutionizing microbiology, as well as having a significant impact on genomic studies of eukaryotes. Complete genome sequence information provides novel insights into genome organization, gene content, and regulation of gene expression. The field of comparative genomics is changing our understanding of evolution, phylogeny, and microbial pathogenesis. Given that microbes represent the bulk of the biomass on the earth, with many unidentified species, it is not surprising that initial genome sequencing efforts sought to survey this biodiversity. However, the large amount of sequencing data being generated underscores just how little we know about microbial species. Among bacterial genomes that have been sequenced, the majority have been bacteria of importance to human health. But, to date, the complete sequence of only one agriculturally important bacterium has been published, the citrus pathogen Xylella fastidiosa (Simpson et al. 2000).
Bradyrhizobium japonicum, the nitrogen-fixing symbiont of soybean (Glycine max), is among the most agriculturally important plant-associated bacteria. B. japonicum is an autochthonous and saprophytic soil bacterium that has the ability to infect the roots of soybean and enter into a symbiotic, nitrogen-fixing association with the host plant. The result of this interaction is the development of a novel plant organ, the root nodule, in which the nitrogen-fixing bacteria reside. The fixed nitrogen produced by this bacterium is subsequently assimilated by the host, and improves plant growth and productivity. Overall, N2 fixation supplies ∼50% of the nitrogen used in agriculture, and because the fixed nitrogen is used directly by the host plant without initial passage through the soil, the process is considered to be environmentally friendly and sustainable (Vance 1998).
We have selected B. japonicum strain USDA110 for genomic studies because a recombination-based physical-genetic map has already been constructed for this strain (Kündig et al. 1993, 1995). USDA110 has also been used extensively for genetic studies and is biochemically and physiologically well defined; in addition, field and laboratory experiments have shown this strain to be highly competitive for nodulation and nitrogen-fixation ability (Lohrke et al. 1996). Furthermore, this strain does not contain plasmids (Sadowsky and Bohlool 1983).
Bacterial genomes have generally been sequenced using a shotgun approach. At 8.7 Mb, the genome of B. japonicum is nearly two to three times the size of most bacterial genomes. In addition, it now appears that a number of genes in B. japonicum may be duplicated (see Results). The large size of the B. japonicumgenome and the possibility of gene duplication makes the development of a physical framework highly desirable to facilitate a shotgun sequencing effort for the purpose of sequence assembly, alignment of sequencing contigs, finishing, and gap closure. A framework approach often involves the development of a large insert library, a contig-based physical map, and a sequence-tagged connector (STC) database.
In most cases, physical maps for bacterial genomes are developed using a restriction-mapping approach with rare-cutting enzymes and pulsed field gel electrophoresis (PFGE) (Romling et al. 1992; Kündig et al. 1993; Irazabal et al. 1997; Riethman et al. 1997). However, these maps do not provide sufficient resolution to act as guides for whole or partial genome sequencing. They also do not provide the resources needed for the immediate cloning of genes involved in key biochemical pathways.
Although contig-based physical mapping is not necessarily a new advance in bacterial genomics, bacterial artificial chromosome (BAC) clone technology has just recently been exploited to develop high-resolution contig-based physical maps for several archael and bacterial genomes (Diaz-Perez et al. 1997; Brosch et al. 1998; Dewar et al. 1998; Tomkins et al 1999). The large-insert BAC system is very appealing as a vehicle for genome analysis in bacteria because the confounding effects of repetitive DNA are expected to be relatively negligible. Therefore, we have employed BAC technology as a tool to develop a physical framework for the B. japonicum genome. The specific goals of the present study were: (1) develop and characterize a BAC library for B. japonicum; (2) develop a high-resolution physical map of the genome consisting of fingerprinted BAC contigs (Soderlund et al. 2000); (3) hybridize mapped molecular markers to the library; and (4) end-sequence enough BAC clones to provide a dense STC database (Venter et al. 1996).
RESULTS
Library Construction and Characterization
A BAC library was constructed for B. japonicum that is suitable for physical mapping and cloning genes associated with plant–microbe interactions. HindIII was used as the cloning enzyme because complete digests with B. japonicum genomic DNA produced fragments ≤30 kb. The library consists of 4608 clones stored in 12 384-well microtiter plates. The number of empty vectors is negligible because all of the 156 clones randomly sampled from the library contained inserts. Figure 1A shows 25 randomly selected clones digested with XbaI to release the insert. We were unable to use the NotI sites in pBACindigo that flank the multicloning site because NotI is a GC 8-base cutter and the B. japonicum genome is relatively GC-rich. Because XbaI is a rare cutting enzyme in B. japonicumand has two unique sites in pBACindigo, digestion of the BAC withXbaI yields a 6.0-kb vector fragment and the BAC insert with 2.0 kb of vector attached. Figure 1B shows a Southern blot of the gel in Figure 1A probed with total B. japonicum genomic DNA; the strong hybridization in all lanes indicates that the source of cloned DNA originated from B. japonicum.
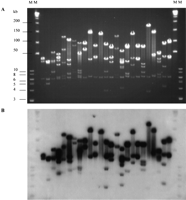
Twenty-five randomly selected Bradyrhizobium japonicum BAC clones digested with XbaI. (A) Ethidium bromide-stained CHEF gel (5–15 sec switch time, 15 h) showing insert DNA above and below the 6-kb pBACindigo XbaI vector fragment band. (B) Autoradiograph of gel in A after Southern transfer and probing with total B. japonicum genomic DNA. Molecular markers (New England Biolabs) are λ ladder (insidelane) and 1-kb DNA ladder (outside lane).
To determine the size distribution of BAC clones in the library, the insert size of the randomly sampled 156 BACs was estimated from theXbaI digests. Based on this analysis, the average insert size is 146 kb, with a range of 30 to 310 kb (Fig.2). More than 78% of the clones are larger than 100 kb. In fact, most of the clones (65%) are between 100 and 200 kb in size. The majority of clones present in the library therefore provide an ideal substrate for physical framework development.
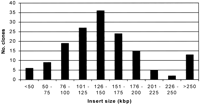
Insert size distribution of clones in the Bradyrhizobium japonicum BAC library. To estimate insert size range, BAC DNA inserts from 156 randomly selected clones were analyzed, as shown in Figure 1A. Results indicate that the average insert size is 146 kb with >78% of the clones >100 kb.
Fingerprinting and Contig Assembly
All 4608 BAC clones were fingerprinted using HindIII. After manual analysis to remove poorly resolved fingerprints, 4441 fingerprints were assembled into contigs using Fingerprint Contig (FPC) software (Soderlund et al. 2000). This initial contig assembly placed 3410 clones into six contigs, with 1032 singletons. The contigs are quite large, containing between 65 and 1206 clones and ranging in size between 250 kb and 2950 kb (Table1). Using PFGE of linearized chromosomes,Kündig et al. (1993) and Jumas-Bilak et al. (1998) estimated thatB. japonicum contained one major replicon of ∼8.7 Mb. The cumulative sum of all six contigs produces an approximate total coverage of 8.68 Mb. The six contigs therefore almost certainly represent the entire B. japonicum genome.
Summary of FPC Contig Development
Contig joining was made possible by hybridizing end probes to high-density BAC colony filters. The RNA end probes were derived by hot amplification of the end sequences from selected BACs representing the ends of each contig. Sufficient overlapping sequence between BACs at the contig boundries allowed for hybridization-based merging of contigs 2, 4, and 6 and contigs 1, 3, and 5, respectively. This resulted in the development of the two large contigs illustrated in Figure3. The FPC analysis did not initially merge the six smaller contigs into the two larger contigs because of a lack of significant overlap in the fingerprints required for the stringent cutoff value that was set to provide optimal accuracy.
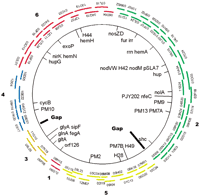
Six FPC contigs illustrated by a selected set of BAC clones providing a complete tiling path of the Bradyrhizobium japonicum replicon. Markers used to orient and anchor the contigs to the genetic map published by Kündig et al. (1993, 1995) are indicated on the inside of the replicon. BAC contig ID numbers are shown on the outside of the replicon and correspond to Table 1. The remaining gaps between the two major contigs obtained via hybridization-based end-joining of FPC contigs 4, 2, and 6 and contigs 3, 1, and 5 are indicated on the inside of the replicon.
BAC End-Sequencing
To develop an STC framework (Venter et al. 1996) and to provide a gene survey of the B. japonicum genome, the ends of 1152 BAC clones were sequenced in both forward and reverse directions. The 2304 sequence traces had an average read length of 726 bases before vector removal. High quality bases are defined as having a PHREDquality value of ≥20, and high quality sequences have ≥100 high quality bases after vector removal. Of 2304 sequence traces, 1939 were high quality sequences (84%), with an average of 709 high quality bases. Compared with the distribution of clones among the six contigs, the high quality STCs appear to be evenly distributed throughout the entire physical map.
Anchoring and Annotation of the Physical Map
An initial genetic-physical map was assembled for the B. japonicum genome consisting of 64 unique markers located throughout the replicon (Kündig et al. 1993, 1995). We selected 14 evenly spaced genes from this map and hybridized them to high-density BAC colony filters, but surprisingly, only five of these markers localized to unique locations in the physical map. Duplication of the other markers suggested the possibility of gene duplication or the presence of gene families. The placement of the 14 unique markers allowed us to anchor and orient the larger of the two contigs, but further marker placement was required to confirm the orientation of both contigs. We obtained all public B. japonicum nucleotide sequences from GenBank and used FASTA (Pearson and Lipman 1988) to compare them to our STC sequences. Positive matches between STCs and known B. japonicum sequences were added to the physical map when >95% sequence identity was observed between an STC and the known nucleotide sequence of a B. japonicum gene. This allowed the unambiguous placement of an additional six B. japonicum markers from the published genetic map. These FASTA-derived markers confirmed the orientation of both contigs.
We were able to obtain 36 additional B. japonicum marker clones that we hybridized to high-density BAC colony filters and added to our physical map. Another 17 unmapped B. japonicumnucleotide sequences from GenBank showed >95% identity to one or more of the STCs in a FASTA search and were also added to the physical map. Finally, we used BLASTX (Altschul et al. 1997) to compare all of our STCs to the nonredundant protein database of GenBank. Using a strict 10−10 E-value cutoff, we found 441 STC hits. From these results, we added 113 unique putative genes to our physical map. The final map contains the general locations of 177 different genes, identified by hybridization, FASTAsimilarity to nucleotide sequences, and BLASTX similarity to protein sequences (Fig. 4). Including the 17 BAC end hybridizations, there are 194 markers on the physical map (1/45 kb). Counting genes only, the physical map contains 177 robust markers (1/49 kb).

Complete physical map with integrated hybridization-based markers and STC sequences. Red text indicates markers from hybridizations and STCs that are known Bradyrhizobium japonicum genes based onFASTA similarity. Black text indicates position ofBLASTX STCs. This map is not to scale and positions shown are approximate.
Functional Characterization of STCs
Using BLASTX to search GenBank at a less stringent cutoff of 10−4, we found 1338 significant hits to our STC sequences. These significant hits represent 69% of the sequenced DNA, a figure that is consistent with our previous STC survey of a bacterial genome (Tomkins et al. 1999). After removing redundancy, the hits that had a minimum 125-base STC sequence (864) were manually evaluated and placed in different protein function categories. A brief summary of the categorized database search results is shown in Table2. The complete BLASTX results are available at the Bradyrhizobium japonicum Genome Project Web site (http://www.genome.clemson.edu/∼twood/projects/brady.html).
Summary of Functional Binning of 864 Nonredundant BLASTX STC Query Results (P < 10−4)
After sorting the sequences by function, the largest group of sequences (40%) is found to be associated with metabolism and biosynthesis. Sequences associated with transport/binding proteins comprised 10.2%. Sequences associated with DNA and protein regulation represented 6.5%. Another 4.7% of the sequences were associated with cell division and DNA replication/repair. Cell structure/membrane sequences were 3.6% and cell signaling sequences represented 1%. Although there can be some overlap between genes that are involved in symbiosis and metabolism, the number of strictly symbiosis-related STCs constituted 1.2% of the dataset. The number of unclassified sequences with GenBank hits totaled 30%. Unidentified and unclassified sequences may represent a subset of those proteins that are specific to B. japonicum and/or proteobacteria in general. The sequencing of proteobacterial genomes is negligible, and they are underrepresented in the public sequence databases.
DISCUSSION
We have successfully constructed a BAC library for the bacterial genome of B. japonicum strain USDA110, an agriculturally important bacterium containing genes involved in nitrogen fixation, symbiosis, and novel enzymatic reactions. The library is an ideal substrate for a number of genomics-based applications, including physical mapping, DNA sequencing, and the cloning of important genes and pathways involved in plant–microbe interactions.
Using this new BAC library, we successfully constructed a highly detailed physical map of the B. japonicum genome by contig assembly via BAC fingerprinting and end probe hybridizations. We were fortunate that in the case of B. japonicum, there were a significant number of molecular markers available that were derived from a previously published recombination-based physical and genetic map (Kündig et al. 1993, 1995). This aided in the alignment, orientation, and verification of the BAC contigs.
It is noteworthy that the hybridization data also allowed the BAC library to be evaluated for genome representation. Positive signals were obtained for all the marker hybridizations with an average of 46 hits per probe and a range of 5 to 95. Although these data indicate preferential cloning of some regions as would be expected using a restriction-enzyme approach to DNA fragmentation, positive signals were achieved in all hybridizations, indicating that the genome was well represented. Furthermore, the number of positive signals obtained by the hybridizing of end probes for the purpose of contig end-joining was even higher, with an average of 69 and a range of 23 to 138.
Using 1152 clones, BAC end-sequencing was performed (forward and reverse) to generate 2256 successful reads for the development of an STC database and preliminary gene survey. There are currently 194 sequences and markers positioned on the B. japonicum physical map to provide a visible high-resolution mapped image of the replicon. The 194 markers comprise clone hybridizations derived from previous recombination-based mapping efforts, the hybridization of contig boundary sequences, the placement of STCs representing known B. japonicum genes, and the placement of 113 unique and robust newly characterized B. japonicum sequences. The 113 newly characterized B. japonicum sequences are derived from 441 STCs in a functionally categorized database derived from high-stringencyBLASTX searches that can be readily traced to a specific map location based on the contig assignment of their respective BACs. As a result, there is now a well-characterized marker every 45 kb throughout the B. japonicum genome.
We have effectively demonstrated the utility of using BAC clones in developing a comprehensive physical framework for a large bacterial genome. These techniques have allowed the construction of a detailed physical map combined with a functionally characterized STC database. This framework approach will provide essential information related to genome structure and organization that will serve as an alignment and finishing tool in the shotgun sequencing of the B. japonicum(USDA110) genome that has recently been initiated (Kasuzu Institute, Japan). Furthermore, this system will provide the necessary tools needed for the cloning and analysis of important genes and whole operons or genetic pathways from other closely related strains that are not being sequenced. When large-scale genome sequencing funds become limiting, this framework approach could be readily applied to any bacteria. Therefore, with a limited budget, an information-rich molecular scaffold can be achieved and utilized for a diverse number of genomics applications.
METHODS
BAC Library Construction
The single-copy BAC library vector, pBACindigo, was obtained from Dr. Hiroaki Shizuya (modified form of pBeloBAC11; Shizuya et al. 1992) and prepared as described by Woo et al. (1994). Megabase bacterial DNA embedded in agarose plugs was obtained as described byRiethman et al. (1997). Partial digests of megabase DNA were performed as follows: chopped plugs were distributed in 100-μL aliquots and incubated on ice for 30 min with 14 μL of 10× enzyme buffer, 14 μL of 40mM spermidine, and 1.4 μL of BSA (10 mg/mL). After a second 30-min incubation with 2 units of HindIII on ice, digestion reactions were allowed to proceed for 20 min at 37°C. Digestions were stopped by placing on ice and adding 1/10 vol 0.5 M EDTA. Partially digested megabase DNA was subjected to an initial size selection by PFGE (CHEF mapper apparatus, Bio-Rad). The first size selection conditions were: 1% agarose, 1–40-sec linear ramp, 6 V/cm, 12°C, 18-h run time, and 0.5× TBE buffer. Three gel slices were excised between 100 and 350 kb and transferred to a second CHEF containing 1% low melting temperature agarose and run at a constant 4-sec switch time under similar time and temperature conditions. Three gel slices were excised, and DNA was removed from the agarose by electroelution using the BIO-RAD Electro-Eluter (Model No. 422) system. Ligations were performed in 150-μL reactions using 30 ng of vector and 250 ng of insert DNA and allowed to proceed for 18 h at 16°C. Desalting was performed via drop dialysis using 0.025-μm (pore size) 25-mm nitrocellulose filters (Millipore) floated on 0.5× TE (10 : 1) for a period of 90 min. After desalting the ligation mixtures, transformations were performed using 2 μL of ligation reaction and 20 μL of competent cells (DH10B, GIBCO BRL). Electroporations were performed in a cell porator with voltage booster (GIBCO BRL) using 320 V at a resistance of 4 kΩ. Transformed cells were diluted immediately with 0.5 mL of SOC (Sambrook et al. 1989) and incubated for 60 min at 37°C before being plated on selective LB (Luria-Bertani) medium with 12.5 μg/μL of chloramphenicol, 0.55 mM IPTG, and 80 μg/mL of X-Gal. After a 20-h incubation at 37°C, the plates were placed at room temperature in the dark for an additional 20 h to allow stronger color development of nonrecombinant colonies. After determining insert sizes of clones, a ligation derived from the 175- to 250-kb gel fraction was used for additional transformations to construct the library. Recombinant white colonies were picked robotically (Genetix Q-bot) and stored individually in 12 384-well microtiter plates (Genetix) containing 50 μL of freezing broth (Woo et al. 1994). After incubation overnight, microtiter plates were stored at −80°C. An initial set of four library replicas was made, and additional copies can be produced upon request (http://www.genome.clemson.edu).
BAC Clone Characterization
To prepare BAC DNA, 3 mL of LB–chloramphenicol (12.5 μg/μL) cultures were grown overnight in 5-cell autogen tubes and miniprepped robotically (Autogen 740 plasmid isolation system). To estimate insert size and determine distribution of clone size, a total of 156 BAC preps were performed from clones selected at random throughout the library. The BAC DNA was digested with 7.5 units (10 h at 37°C) ofXbaI and analyzed by PFGE in 1% agarose gels (6 V/cm, 5–15-sec switch time, 15-h run time, 14°C). Southern blots of size-separated BAC inserts were performed using standard protocols (Sambrook et al. 1989) after UV-nicking the gels (Gene Linker, Bio-Rad).
BAC Library Screening
High-density colony filters for hybridization-based screening of the library were prepared using the Gentix Q-Bot. Clones were gridded in double spots using a 3 × 3 array on 11.5 × 22.5-cm Hybond N+ filters (Amersham). This gridding pattern allows 4608 clones to be represented per filter. Colony filters were grown and processed using standard techniques (Sambrook et al. 1989). For hybridizing gene probes, clone inserts were cut from ethidium bromide-stained agarose gels and DNA was extracted using a QIAEX II gel extraction kit (QIAGEN). Radiolabeling of probe DNA and hybridization of colony filters were performed using standard techniques (Sambrook et al. 1989) with the following modifications: hybridizations were performed for 16 h at 65°C and filters were washed twice at 65°C (15 min per wash) in 1× SSC/0.1% SDS solution. Preparation of single-stranded radiolabeled RNA probes from the end sequences of BACs was done according to manufacturer's instructions using the Strip-EZ RNA T7/SP6 Kit (Ambion).
BAC Fingerprinting and Contig Assembly
BAC DNA for fingerprinting was prepared as described previously for BAC clone characterization. The process of fingerprinting, digitizing of gel images, and contig building has been described previously (Marra et al. 1997). The IMAGE software program version 3.5 (Sanger Centre, UK) was used to digitize gel images. All bands were manually checked, and bands below 1 kb were ignored because they were not well resolved and accounted for <1% of the total length of the BAC inserts. The Fingerprint Contig (FPC) software program version 4.6 (Sanger Centre, UK) was used for contig building. Initial contig assembly was done at high stringency with a tolerance of 7 and a cutoff score of 10−13. These threshold values were chosen to minimize error and maximize contig size and were determined empirically for this data set. After marker data were collected and added to the map, contigs were merged at a lower stringency of 10−8.
BAC End-Sequencing
To prepare BAC DNA for end-sequencing, 4 mL of LB–chloramphenicol (12.5 μg/μL) was inoculated with 4 μL of BAC freezer stocks, and cultures were grown for 20 h in 6-cell autogen tubes at 37°C. BAC cultures were then miniprepped robotically (Autogen 740 plasmid isolation system), and DNA was resuspended in 25 μL of 1 mM Tris. Sequencing reactions were set up according to manufacturer's instructions for the Big Dye Terminator chemistry (Perkin Elmer). Samples were then loaded onto 48-lane sequencing gels in ABI377 automated sequencers. Gels (250 mL) were composed of the following: 5% Long Ranger (FMC), 6 M urea, TEMED 18 μL, 150 μL of ammonium persulfate (10% stock), and 1× TBE buffer. Reaction products were electrophoresed using a 3.5-h run. Base-calling was performed automatically using phred, and vector sequences were removed by CROSS_MATCH (Ewing and Green 1998; Ewing et al. 1998). High quality end-trimmed BAC end sequences (defined as those having >100 nonvector bases with a PHRED quality value >20) were used as queries in FASTA andBLASTX searches of the GenBank database. All software was run locally on a Sun Ultra30 workstation using Solaris 2.6. The sequence data described in this paper were submitted to GenBank under accession numbers AZ933551–AZ935555. Detailed mapping and sequencing data are available at the B. japonicum Genome Project Web Site (http://www.genome.clemson.edu/∼twood/projects/brady.html).
Acknowledgments
Appreciation for the provision of B. japonicum gene probes is extended to Eulogio J. Bedmar, Arvind Bhagwat, Mary Lou Guerinot, Elmar Kannenburg, Peter Mueller, Mark O'Brian, Eric Triplett, and Peter van Berkum. Thanks to Heather Miller-Smith, Michael Atkins, and John Bishop for providing technical assistance during library replication and filter development. Thanks to Cari Soderlund (CUGI) for assistance with the FPC software program. Thanks also to Stephanie Higgingbottom and the physical mapping group at CUGI for providing assistance during the fingerprinting phase of the project. This research was supported by a Clemson University Research Grant Committee grant to J.T., Coker Endowment (Clemson University) and NSF MRI grant no. 9724557 to R.W., USDA-NCRICGP grant no. 99-35305-8323 and University of Tennessee Graduate School Faculty Research Award to G.S., and University of Minnesota Agricultural Experiment Station funds to M.S.. BAC library resources (library, clones, and high-density colony filters) for B. japonicum can be ordered at the CUGI Web site (http://www.genome.clemson.edu).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL jtmkns{at}clemson.edu; FAX (864) 656-3443.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.185001.
-
- Received February 16, 2001.
- Accepted May 14, 2001.
- Cold Spring Harbor Laboratory Press








