
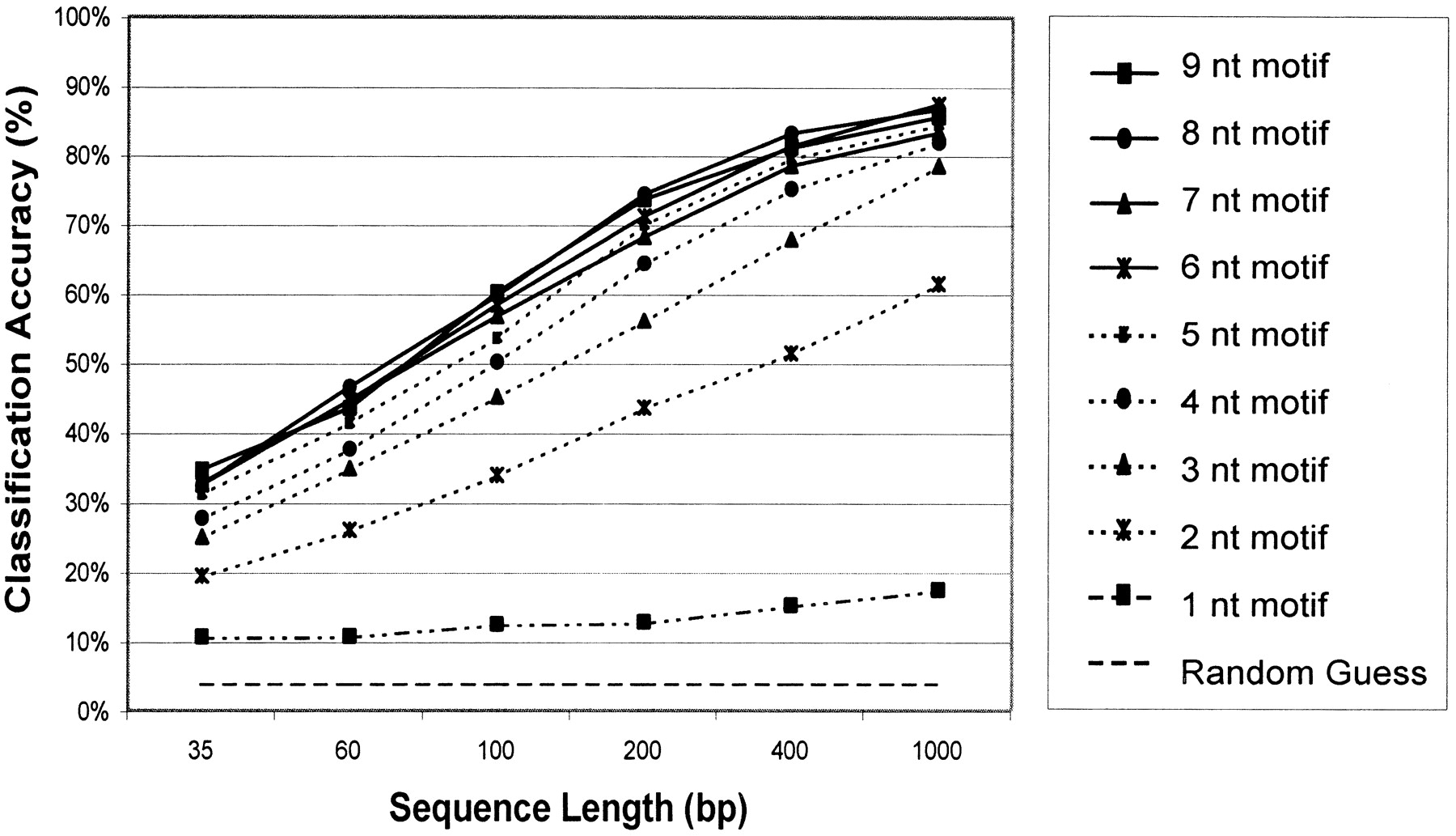
Dependence of classification accuracy upon motif and sequence length. Motif lengths ranging from one to nine base pairs were evaluated for classification accuracy. A default (“random guess”) classifier is also shown. For each motif length, six different sequence lengths were tested. (35, 60, 100, 200, 400, and 1000 bp). The classification accuracy in percent is represented on they-axis as the arithmetic mean over the independent genome results. One hundred sequences were randomly picked from each genome for each sequence length, and the classification accuracy was calculated as the ratio of correct predictions, divided by the total number of predictions for each genome and test runs. On thex-axis, the different sequence lengths are shown. Training was performed on 90% of the genome sequences (“training set”), and the remaining 10% (“test set”) was used to evaluate classification accuracy.








