
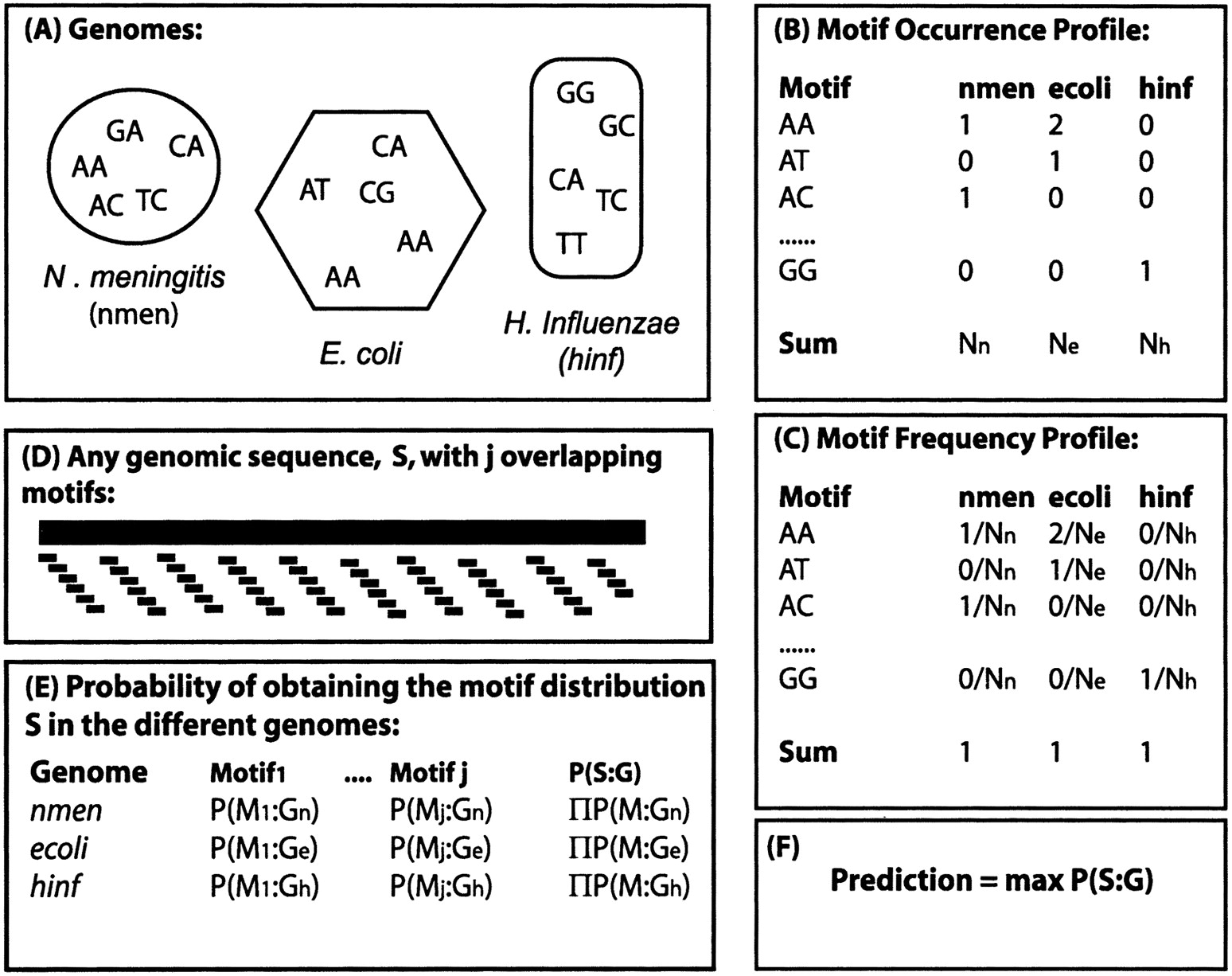
Outline of the Bayesian classifier. (A) For a given motif length (in this figure, two base pairs), the occurrence of all overlapping motifs for each genome is recorded in the motif occurrence profile (B). The motif occurrence profile for each genome is then transformed to a motif frequency profile (C) by dividing each motif occurrence by the total number of motifs in that genome. (D) A sequence, S, of arbitrary length is taken at random from any of the genomes, consisting of a number of j overlapping motifs. (E) The probability of obtaining the motif distribution present in sequence S is separately calculated for each genome and motif. For example, the probability of obtaining motifi in E. coli, P(Mi:Ge) is estimated by the frequency of that motif in the E. coligenome, calculated in (C). The probability of obtaining the motif distribution present in sequence S is then estimated as the product of the individual probabilities of obtaining each motif (E). The classifier predicts the most probable genomic origin (F), the genome with the highest probability P(S:G).








