
Identifying Functional Elements by Comparative DNA Sequence Analysis
Functional elements in DNA sequences tend to evolve at a much slower rate than nonfunctional sequences, because functional elements are subject to selective pressure. Comparative DNA sequence analysis exploits this local difference in mutation rates to identify functional elements such as genes, regulatory sequences, splice sites, and binding sites. This is accomplished by comparing orthologous sequences from two or more species and identifying those regions that are most well-conserved across the species. These conserved regions are excellent candidates for further experimentation. Such a comparative analysis is sometimes called “phylogenetic footprinting” (Tagle et al. 1988; for review, see Duret and Bucher 1997).
In this issue, Cliften et al. (2001) report the results of one such comparative study. Their study, focusing on the yeastSaccharomyces cerevisiae, had two separate goals. The first was to use orthologous sequences from seven other partially-sequencedSaccharomyces species to predict short protein-coding genes, RNA genes, and regulatory sequences, all of which create challenges for current computational tools. The second was an investigation into which combination of these species, if completely sequenced, would be likely to shed the most light onto future comparative studies of the genus.
When selecting species for comparative sequence analysis, one challenge is that the species should be sufficiently diverged that functional elements stand out from less-conserved nonfunctional sequence, yet sufficiently close that (1) the orthologous functional elements have not been lost in evolution, and (2) alignment algorithms such asBLAST (Altschul et al. 1990) and CLUSTALW(Thompson et al. 1994) will correctly align those orthologous elements. The issue in alignment is that functional elements such as regulatory sequences can be quite short compared to the surrounding nonfunctional sequence. In this case, the noise caused by aligning the diverged nonfunctional sequences may well cause the functional elements to remain unaligned and undetected.
Cliften et al. (2001) chose four Saccharomyces species from the sensu stricto group, two species from the sensu lato group, and one petite-negative species. They sequenced >4.3 Mb from these genomes and used the resulting sequences in their comparative studies. The authors discovered interesting new candidate genes and regulatory sequences, and also drew conclusions about the choice of species for future studies. They generally found the sensu stricto species to be too close to S. cerevisiae to allow discrimination between functional and nonfunctional elements, and the other species too distant for accurate alignment to S. cerevisiae. To overcome these problems, they recommend using a combination of at least four species simultaneously and, when alignment algorithms fail, using a motif discovery algorithm such as AlignACE (Roth et al. 1998).
This idea leads us to recent advances in phylogenetic footprinting methods (Blanchette et al. 2000; Blanchette 2001) that allow the use of quite diverged species, by abandoning alignment altogether. These studies introduced specialized phylogenetic footprinting algorithms that find the most conserved motifs among the input sequences, as measured by parsimony score on the underlying phylogenetic tree. The algorithms were used successfully to identify a variety of regulatory elements, some known and some novel, in sets of diverse vertebrate DNA sequences as well as in sets of diverse plant DNA sequences.
Figure 1 illustrates an example fromBlanchette (2001) of binding sites predicted by the algorithm when applied to the 5′ untranslated region of the plant rbcS gene. All three boxes shown are known regulatory elements for this gene (Arguello-Astorga and Herrera-Estrella 1998). The 10 plants shown span ∼760 million years of evolution. The regulatory elements found are each only nine basepairs in length, so (not surprisingly) multiple alignment algorithms such as CLUSTALW fail to align these elements. Note in Figure 1 that the phylogenetic footprinting algorithm identifies multiple occurrences of the I-box in some of the regions and also identifies the G-box even though it is missing in the monocotyledons.
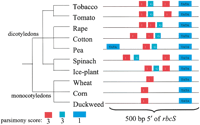
Predicted binding sites in the 5′ untranslated region of the plantrbcS genes. Distances are approximate.
Even if such specialized footprinting algorithms obviate some of the need for alignment, studies of which genomes to compare—such as that of Cliften et al. (2001)—remain important. One reason for this is that the species must still be chosen carefully, so that the interesting functional elements are conserved and the nonfunctional sequences are not.
With the number of genome projects completed and underway, the coming years promise exciting discoveries through such phylogenetic footprinting studies. An important part of this endeavor will be the development of algorithmic methods designed specifically for such comparative studies.
Footnotes
-
E-MAIL tompa{at}cs.washington.edu; FAX (206) 543-8331.
-
Article and publication are athttp://www.genome.org/cgi/doi/10.1101/gr.197101.
- Cold Spring Harbor Laboratory Press








