
Directed Gap Closure in Large-Scale Sequencing Projects
Abstract
A problem in many sequencing projects is the final closure of gaps left in the clone libraries, which serve as templates for sequencing, because of uncloned or unclonable genomic areas. By use of theXylella fastidiosa genome as a test system, we present here an approach to generate, in a directed manner, sequence information from those gaps. We suggest using the complete clone library as a competitor against the genomic DNA of interest in a subtractive hybridization procedure similar to representational difference analysis (RDA). The resulting sequence information can be used to screen selectively other clone resources or serve directly for gap closure.
One major task in genome sequencing is the gap closure subsequent to the high throughput sequencing phase. Cloning gaps that remain as a result of biological factors or statistical cloning fluctuations prevent successful cloning of the respective sequences. Examples of tedious gap closing efforts are numerous both in shotgun approaches and in strategies that are based on pre-ordered clone libraries, in micro-organisms as well as in higher eukaryotes (e.g.,Fleischmann et al., 1995; The C. elegans Sequencing Consortium 1998; Adams et al. 2000; The Chromosome 21 Mapping and Sequencing Consortium 2000) The parallel use of two or more types of libraries seems to be at least a partial solution. Smaller gaps might be closed by PCR, if information concerning the contig ends is available. However, this is not possible for large gaps. Here we present an approach to obtain sequence information almost exclusively from such gaps. On the basis of representational difference analysis (RDA) (Lisitsin et al. 1993; Hubank and Schatz 1994), we applied a simplified protocol of one subtractive hybridization followed by PCR to enrich for sequences, which are not represented in the respective clone library. As a test system, we used a library of 1051 cosmid clones covering the 2.7 Mb of the Xylella fastidiosa genome (Frohme et al. 2000). This library was used in the X. fastidiosa genome project (Simpson et al. 2000) providing the template for the bulk of the genome sequence. Although the library had a 15-fold coverage, 13 regions ranging in size between ∼700 bp and >40 kb were not present, totaling 208 kb (∼8%) of the genome. These gaps were eventually closed with the help of PCR, extra λ clones and random sequences obtained for the entire genome.
RESULTS
For the selective isolation of gap sequences, we used two DNA populations. The tester was prepared from X. fastidiosagenomic DNA that was digested with Sau3AI or BamHI, respectively, and ligated to PCR adapters for later amplification. For the driver, the whole cosmid library was used after digestion with the same restriction enzymes. A 2000-fold excess of driver was used in the subtractive hybridization experiments. The subsequent PCR resulted in difference products enriched for fragments, which originated from the gap areas.
Gel electrophoresis of the difference products showed a smear ranging from 100 to 900 bp for the Sau3AI-digested DNA, and discrete bands between 500 bp and ∼2 kb for the BamHI-digested DNA (Fig. 1) as one would expect, because the enzymes have 4-bp and 6-bp recognition sites, respectively. A total of 314 clones from both the Sau3AI and the BamHI preparation were picked and end-sequenced. Taking into account redundant information, the 314 sequences could be reduced to 168 sequence clusters, of which 159 contained X. fastidiosa DNA. Of these, 141 clusters contained only one or two sequences, whereas 27 clusters consisted of 3 to 26 sequences. The latter resulted mainly from the experiment with BamHI. The nine remaining clusters without X. fastidiosa sequence were of unknown origin and showed only low homology to any known sequence stored in databases.
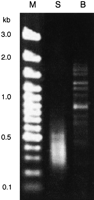
Gel separation of the difference products. S and B indicate the enzymes (Sau3AI and BamHI) used for the initial restriction digest. (M) 100-bp marker ladder (MBI Fermentas, Germany).
Of clones that represent the 159 X. fastidiosa clusters, 123 (74% of the total) had an insert DNA which is not contained in the cosmid library but is part of the final X. fastidiosa genome sequence. A detailed comparison showed that with the exception of two tiny gaps of ∼0.7 and 1.3 kb, all other gaps were represented by sequences in our gap-filling library (Fig.2). Moreover, the 1285-bp long X. fastidiosa plasmid (pXF1.3), which is not present in the cosmid library, was represented by three fragments covering more than half of the plasmid's sequence. The end sequencing provided more than 28 kb of gap-specific sequence information, with individual read lengths of up to 700 bp. In reality, the clones contained more gap sequence, of course, that was not accessed, however, because only end-sequencing was performed. Moreover, further clone picking and sequencing would have produced significantly more gap-specific sequences, because the redundancy observed in the library resulting from the Sau3AI experiment was quite low.

Representativity of gap-filling sequences. The lower row of blocks represents position and length of the gaps in the Xylella fastidiosa cosmid library as determined elsewhere (Frohme et al. 2000). Bars in the top row indicate the location and distribution of 123 gap-specific fragments within the 2700-kb X. fastidiosa genome.
DISCUSSION
The approach presented here provides the means to produce, selectively, sequence information for an accelerated gap closure in genomic sequencing projects. The experimental procedures involved are simple to perform and provide a high proportion of gap-specific sequences. Restriction enzymes with both 4- and 6-bp recognition sequences were found to be useful. Usually, the fragments resulting from a digest with the former can be sequenced completely by a single read. The overall larger fragments produced by the latter enzyme might be a more efficient choice for the closure of wider gaps, because the sequence redundancy could be reduced by using them as templates in a primer walking strategy. In any case, however, the resulting fragments do not only provide sequence information but are also helpful starting points for other approaches of gap closure. The sequence information is useful for the generation of PCR or sequencing primers located within gaps, for example, or for the generation of probes to be hybridized to other library types to identify potentially gap-spanning clones. In this last respect, a labeled difference product may be used directly as a probe for screening another library.
The relatively high portion of false positives — one quarter of the clones did not contain gap-specific Xylella DNA — is probably the result of the single step of subtraction enrichment, done in an attempt to reach a compromise between specificity and representation. Further enrichment steps, as are usually performed in conventional RDA experiments, would probably have resulted in a smaller number of unwanted non-gap sequences due to a stronger selectivity. However, it could have caused a bias toward fewer fragments, resulting in a less complex DNA population.
As in all techniques that enrich DNA fragments by subtraction, external contamination could be a problem in this gap-filling procedure. However, the enrichment factor is considerably lower in this setup than in conventional RDA. Therefore, contamination is less of a problem. Nevertheless, all precautions routinely adopted for highly sensitive, PCR-based protocols were applied, particularly during the preparation of the starting material for the tester. In addition to potential contamination, we experienced another technical problem: Chimeric clones can occur, if a low ratio of adaptor:DNA is used during the ligation at the very start of tester preparation. Thus, a careful determination of the DNA concentrations is critical.
Although the results presented here are based on a genomic cosmid library, the procedure should be applicable irrespective of the type of vector system that is being used. At an earlier stage of the project, only a subset of (then) sequenced cosmids had been used in a preliminary experiment. As expected, a more complex fragment pattern could be seen in the gel electrophoresis. These difference products, however, were not analyzed in more detail, because the sequencing had proceeded further. In shotgun sequencing projects, plasmid libraries form the basis of analysis. Because the clonability is usually better for shorter inserts, one would expect fewer real gaps in such projects. Still, the procedure should be useful nevertheless, even if only for a verification of the completeness of the clone coverage.
METHODS
For the tester preparation, few micrograms of X. fastidiosa genomic DNA imbedded in agarose plugs were used. After equilibration with water, the blocks were treated with 5 units of agarase (Roche, Germany) per 100 mg agarose. Subsequent restriction digestion was with either 40 units of Sau3AI or 40 units ofBamHI for 2 h at 37°C, followed by a heat inactivation step. Further purification of the DNA and addition of the adapter molecules followed the protocol of Hubank and Schatz (1994).
For driver preparation, the whole cosmid library was plated on 2YT agar with 30 mg/L kanamycin and grown overnight. The colonies were wiped off with a rubber scraper and used to inoculate a 500-mL liquid culture, which was grown at 37°C to a density of 1.2 units OD600. Plasmid DNA was isolated with the maxi-preparation kit of QIAGEN (Hilden, Germany). A total of 100 μg DNA were digested with 400 units ofSau3AI or BamHI for 4 h and used as driver.
A 5-ng tester and 11.5-μg driver (molar ratio in X. fastidiosa DNA 1:2000) were used in a subtractive hybridization as described (Hubank and Schatz 1994) with a duration of 60 h. The material was amplified by 30 PCR cycles, without any prior mungbean-nuclease digest, resulting in an exponential amplification of tester:tester duplexes only. An initial test PCR was used to determine the linear range of the reaction.
The products of the amplification were directly cloned into pGEM-T-easy vector (Promega, Germany). Insert sequencing followed standard procedures on ABI-377 machines. For the definition of sequence clusters, the SeqmanII software (Lasergene, Germany) was applied. Sequences were compared with the final version of the X. fastidiosa genome and the database of cosmid sequences produced within the genome project (http://www.lbi.dcc.unicamp.br/services/index.html) by use ofBLAST algorithms. The database contains data from 117 sequenced cosmids. Extensive analysis of the library (Frohme et al. 2000), had shown that these cosmids are representative for the entire library of 1051 clones.
Acknowledgments
This work was supported by Federation for the Advancement of Sciences, Sâo Paulo within the Xylella fastidiosa genome project. We thank Joao Carlos Setubal and the bioinformatics group (Campinas SP, Brazil) for their support.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL m.frohme{at}dkfz-heidelberg.de; FAX 49-6221-42-4682.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.179401.
-
- Received January 8, 2000.
- Accepted February 27, 2001.
- Cold Spring Harbor Laboratory Press








