
Segmental Duplications: What's Missing, Misassigned, and Misassembled—and Should We Care?
For many people, the announcement of the release of working draft sequence of the human genome was the climax of more than 15 years of planning and preparation (International Human Genome Sequencing Consortium 2001). Despite the controversy and sensationalism, it was an awesome achievement, culminating in the “genome party of the century”. There was much to celebrate. The majority of genes were identified, mapped to their appropriate location, and await the ascription of phenotypic data.
Among the public, however, there is the impression that the task is a fait accompli. In my case, several family members contacted me after the media blitz to inquire whether I was now out of a job—after all, the Human Genome Project is entering its projected two-year twilight. Indeed, this may be the appropriate time for sequencers and sequence-gazers alike to “jump ship” or at the very least to look beyond the next horizon. The genomic revolution will now launch the proteomics revolution with its promise of tailor-made therapies for the masses. Association studies using SNP data are expected to provide insight into the molecular etiology of complex genetic diseases (Chakravarti 2001). Comparative sequencing of the genome of model organisms such as the mouse and the rat will be used to discover elements critical in the regulation of our own genes and provide an invaluable resource for future mutagenesis studies (Nadeau et al. 2001).
As scientists, we of course know that much work still remains to be done before the final declaration of a finished human genome. We all recognize that gaps remain in the project, and most of the community is committed to rolling up their sleeves and getting on with the final sequence and analysis. Nevertheless, despite this commitment, there remains the impression that gap closure will be akin to “mopping up the dance floor after the band has gone home”; it will be an arduous task with little reward, done by a few people willing to don the overalls, put the trash where it belongs, and pick up the pieces.
Currently, two types of gaps are recognized within the working draft sequence (Bork and Copley 2001). There are gaps that are contained within the sequence assembly of the ordered clones. These are trivial gaps, each no more than a few 100 bp in length. Most will be closed during the “topping-off” of sequence from existing projects. Gaps between ordered clones and sequence contigs are the second type of gap. These are larger in size and potentially more problematic in nature. Some of these will be easily closed by the identification and sequencing of bridging clones obtained from paired-end sequence data. Others represent genomic segments not present within existing clone libraries. Such regions were highlighted during the closure of chromosome 21 and 22 (Dunham et al. 1999; Hattori et al. 2000) and purportedly are similarly recalcitrant to subcloning. Specialized technologies are required to close such gaps in the clone map.
I would like to propose a third type of gap that may be underestimated at present. These are gaps associated with nearly identical sequence segmental duplications. These gaps result from the underrepresentation and misassembly of duplicated sequences in the human genome. Such gaps are particularly onerous because their resolution requires that the duplicated nature of the segments be first recognized and then the suboptimal assembly be untangled.
As part of the International Human Sequencing Consortium, we examined the distribution of nearly identical sequence (90–98% sequence identity and >1 kb in length) duplications throughout the genome and the quality of sequence assembly within such exceptional regions (Bailey et al. 2001; International Human Genome Sequencing Consortium 2001). The analysis revealed that a modest fraction of the genome (∼5%) consists of large duplicated segments often containing complete or partial copies of genic material. The amount of duplication seen is more than most scientists would have anticipated. The size (>10 kb), the fraction, and the degree of sequence identity of these segmental duplications are “unique” attributes of human genome structure. The amount of comparable duplicated sequence within invertebrate genomes is at least an order of magnitude reduced. What is more surprising than the amount, however, is the distribution pattern of this material. Many believed that nearly identical sequence duplications would be restricted to clusters (tandem arrays of genes) or atypical regions of the genome such as pericentromeric/subtelomeric regions and the Y chromosome. Such regions are indeed enriched (8–10-fold) (Bailey et al. 2001). In some cases, blocks of duplications are arranged in a mosaic fashion with individual units comprising larger genomic domains that span multiple Mb of sequence. Such areas, however, only account for one-third of the duplicated blocks. The remainder is dispersed throughout euchromatic and/or gene-rich regions. This organization suggests that the human genome is evolutionarily much more malleable and that paralogous segments are more widely distributed than anticipated. These findings have some serious implications for the assembly of human genome sequence.
There are three possible outcomes when large nearly identical duplicated sequences are encountered during sequence and assembly. (1) The sequences may be recognized as distinct and properly resolved as separate loci, (2) the sequences may be underrepresented due to the presence of virtually identical sequence already in the database, or (3) distinct paralogous loci may be mistakingly assembled into a single sequence contig (Fig. 1). The latter two outcomes, by definition, create gaps. The first of these two is complicated by the fact that duplicated sequences may be particularly difficult to assign due to their multi-site distribution. Not surprisingly, when sequence contigs containing duplicated sequence were examined, we found them much more likely (seven times) to be assigned either to a random location or to an unknown chromosome. Comparison between FISH localization and in silico data for clones harboring duplicated sequences allowed us to crudely estimate other mapping and assembly parameters of paralogous loci (Bailey et al. 2001; Cheung et al. 2001). When duplicated contigs were assigned by FISH, they were often (∼50%) mapped to positions that were discordant with the in silico data. In addition, a significant number of signals (∼30%) for multi-site clones could not be identified by analysis of the working draft, indicating that these regions were underrepresented in the current assembly. Further, many of these contigs did not bridge (by sequence or by paired-end data) into unique sequence regions, indicating that the boundaries of the duplicated sequence had yet to be resolved. An analysis of several highly duplicated regions whose organization had been previously experimentally validated (16p11, 2p11, 19p12, 16p13, and 15q11-q13) found that the current assembly, in most cases, did not recapitulate the organization published in the literature. The most common error was the merging of nearly identical sequence duplicated segments into a single contig.
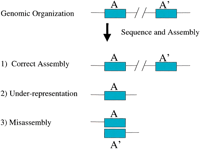
Duplicated sequence and human genome sequence assembly. Three possible outcomes are shown for duplicated sequences A and A'.
The presence of sequence gaps within duplicated regions is not ostensibly a result of current limitations in cloning technology. Rather, the complex, highly duplicated nature of these regions is not amenable to high-throughput assembly methods without further refinement. Within the last two years, output from the human genome project has scaled exponentially. This change represented a fundamental shift in strategy that included the adoption of an intermediate unfinished sequencing product and the selection of clone reagents based on STS assignment and/or fingerprint map data (http://genome.wustl.edu/gsc/human/human_database.shtml) (McPherson et al. 2001). The absence of unique sequence within these regions over large distances (100's of kb) severely biases against the selection of such BACs as templates for sequencing based on STS-PCR (Eichler 1998). Further, BACs that contain large, nearly identical sequence duplications are likely to coalesce into a single fingerprint, making it impossible to distinguish fingerprints from different paralogous loci. One solution to the problem might be to identify these fingerprint contigs that contain too many nearest neighbors and use paralogous sequence variants (PSVs) derived from monochromosomal material to categorize distinct loci (Horvath et al. 2000). The distinction between allelic and paralogous sequence variation is critical particularly in regions where the degree of sequence identity approximates 99%; discernment at the sequence level provides the greatest sensitivity in this regard. Such characterization of BAC clone resources will foster increased coverage in these regions, reduce suboptimal assembly, and concomitantly eliminate gaps in the sequence.
Considering the additional effort that will be required, what possible incentives are there for finishing these regions? The most difficult gaps to resolve will be those located within duplicated segments that are large and nearly identical at the sequence level. Because of the limitations of the working draft sequence, it is currently impossible to estimate what fraction of the genome is duplicated at >98% sequence identity. I would argue that these most elusive targets are the most important. I will make two predictions:
Recent Genomic Duplications Underlie Many Uncharacterized Human Diseases
Over the last 10 years, it has repeatedly been shown that the presence of large blocks of homologous sequences (duplications) flanking unique gene sequences can predispose to recurrent chromosomal structural rearrangements associated with disease (Mazzarella and Schlessinger 1998; Ji et al. 2000). The high degree of sequence identity among nearly identical duplicated copies promotes misalignment of chromosomes during meiosis where recombination occurs among paralogous instead of allelic loci. Consequently, unique sequence in the vicinity of these duplications are deleted, duplicated, or inverted. These genomic imbalances of dosage sensitive/imprinted genes have been shown to result in a variety of childhood diseases (velocardio-facial/DiGeorge, Prader-Willi/Angelman Williams-Beuren Syndrome, etc). How common are these de novo rearrangements? The combined incidence of duplication-mediated childhood diseases has been estimated at ∼1 / 750. Considering the large number of novel nearly identical sequence duplications that have been uncovered during the analysis of the human genome, a much more significant impact on human health should be anticipated. Once all the nearly identical sequence duplications and their associated unique flanking sequences are identified, genome-wide screens using methods such as comparative genomic hybridization may begin to estimate the true incidence of such disease in the human population.
Recent Genomic Duplications are the Engines of Hominoid Evolution
Genome duplications are one of the primary forces of evolutionary change. Duplicate copies of genes through mutation and natural selection can diversify protein function. In most species where new/specialized gene functions have been documented, the changes inevitably have occurred in concert with a duplication event (Nurminsky et al. 1998; Zhang et al. 1998; Duda and Palumbi 1999). Further, most of the discernible events were recent in origin. Is it possible that regions that have recently duplicated in the human lineage were critical in the emergence of our species? There is some evidence that recent duplicated segments may harbor genes that are radically different between us and our closest relatives (Courseaux and Nahon 2001). Although the concept of evolution of human-specific genes may be heretical, the abundance of recently duplicated material and the importance of duplication in evolutionary paradigm justify its consideration. Over longer periods of evolutionary time, the duplication and transposition of genomic segments, could facilitate the juxtaposition of groups of exons from diverse genes. These new combinations could, in theory, lead to the formation of larger genes with more complex functions. One of the major conclusions of the genome sequence papers was that it is not gene number, but rather the complexity of protein modules that distinguishes our genes from those of the invertebrates. Segmental duplication is one way in which domain accretion may have been achieved, by allowing larger genes to grow in a modular fashion.
In short, exceptional duplicated regions underlie exceptional biology. Consequently, I look forward with great anticipation to the unabridged version of the human genome. As the clean-up crews descend onto the genome to produce a finished product, we should dispel notions of mindless drudgery. Although its completion will unlikely be greeted with same level of fanfare, closing the gaps should be heralded as the next major challenge of the Human Genome Project. A finished human genome must be the highest priority—not simply because this was the original intent but because of the remarkable biomedical impact that it will offer (Collins et al. 1998). Considering the unexpected architecture of our genome, the two-year timeline for completion may be overly optimisitic, particularly within the duplicated regions. True finishing will require much more than simply “topping-off” the working draft sequence. A greater investment is necessary to fully resolve the paralogous nature of the human genome. Despite the milestones that have been achieved, the most substantive insights into the organization, evolution and pathology of our genome await discovery.
Footnotes
-
E-MAIL eee{at}po.cwru.edu; FAX (216) 368-3432.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.188901.
- Cold Spring Harbor Laboratory Press








