
Sleepy Dogs Don't Lie: A Genetic Disorder Informative About Sleep
Molecular genetic approaches have facilitated our understanding of numerous diseases and provided important insights into the neurobiological basis of various behaviors. Until recently, the complex behavioral and physiological adaptation known as sleep has been impervious to such approaches. In the last two years, however, a familiar theme has been played out in the sleep field: The discovery of the genetic basis of an animal model of a human disorder has implications both for the treatment of patients and for the neural basis of a specific behavior. In this issue, the paper by Hungs et al. provides the latest chapter in this fascinating and rapidly unfolding story.
Narcolepsy in Humans
Narcolepsy, sometimes called Gélineau's disease after the neurologist who coined the term, was first described more than a century ago (Westphal 1877) and has been of continuing interest because of its unique sleep phenotype. The “narcoleptic tetrad” of symptoms include excessive daytime sleepiness (EDS); cataplexy, a sudden loss of muscle tone during wakefulness usually precipitated by strong emotional stimuli; sleep paralysis, a transient inability to move upon awakening that can persist as long as 15 minutes; and hypnogogic hallucinations, vivid dreamlike imagery at sleep onset that can be quite frightening to a narcoleptic person. The last three symptoms are similar to characteristics of normal rapid eye movement (REM) sleep, a stage of sleep during which dreams are often reported and the voluntary musculature is actively inhibited resulting in muscle atonia. Because of these similarities and the fact that the latency between sleep onset and the first REM period is greatly reduced in narcoleptics, narcolepsy has been thought to be a disorder of REM sleep.
The mode of inheritance of narcolepsy is complex: Although 1%–2% of the first-degree relatives of narcoleptic patients manifest the disorder (compared with 0.02%–0.18% of the general population), the low degree of concordance between monozygotic twins indicates an interaction between environmental factors and a specific genetic background. A link with human leukocyte antigen (HLA) Class II antigens was first established in the 1980s (Juji et al. 1984). Across various ethnic groups, > 85% of all narcoleptic patients with definite cataplexy share a specific HLA allele, HLA DQB1*0602 (most often in combination with HLA DR2), compared with 12%–38% of the general population (Mignot 1998). Such close association with the HLA system has led to the suggestion that narcolepsy may be an autoimmune disease (Langdon et al. 1986).
Narcolepsy in Dogs
Narcolepsy has been described in other mammals including several breeds of dogs (Dachshunds, Poodles, Labrador Retrievers, and Doberman Pinschers), miniature ponies, and Brahman bulls. Although the mode of inheritance in small-breed dogs is as complex as in humans, large-breed dogs such as Doberman Pinschers and Labrador Retrievers provide a simplified genetic system in which the mutation in the canine narcolepsy (canarc-1) gene is transmitted as an autosomal recessive trait with full penetrance. It has been known for some time that canarc-1 is not associated with the dog leukocyte antigen system (Dean et al. 1989; Wagner et al. 2000). The identification of the canarc-1 mutation as a deletion of the hypocretin receptor 2 (Hcrtr2) gene through positional cloning (Lin et al. 1999) was a major advance in this field and suggested a link between the newly-described hypocretin system and sleep. Importantly, the mutation in these two breeds is in a different region of the same gene,Hcrtr2 (= OX2R), both of which result in a truncated, nonfunctional protein. In their present paper, Hungs et al. (2001) identify a Hcrtr2 mutation in a Dachshund family in which the protein is appropriately localized to the cell membrane but fails to bind ligand, and little calcium is mobilized upon receptor stimulation.
The Hypocretin/Orexin Ligand-Receptor System and Sleep
The hypocretin/orexin (Hcrt/Ox) gene was described in 1998 by two groups working independently (de Lecea et al. 1998; Sakurai et al. 1998) and encodes two neuropeptides, alternatively called hypocretin-1 (Hcrt1) or orexin-A (Ox-A) and hypocretin-2 (Hcrt2) or orexin-B (Ox-B). The cell bodies which make the Hcrt/Ox peptides are found in a very restricted region of the posterior hypothalamus but widespread projections from these cells innervate many brain areas, particularly those implicated in the control of sleep and wakefulness (Peyron et al. 1998; Kilduff and Peyron 2000). Two receptors for these peptides are known to date: The hypocretin-1 receptor, Hcrtr1(= OX1R), binds the two peptides with equal affinity whereas Hcrt1/Ox-A binds to the hypocretin-2 receptor,Hcrtr2(= OX2R), at higher affinity than Hcrt2/Ox-B (Sakurai et al. 1998). The two peptides have both direct excitatory (de Lecea et al. 1998) and neuromodulatory (van den Pol et al. 1998) effects on receptors in both the hypothalamus and other brain areas (Hagan et al. 1999; Horvath et al. 1999; Ivanov and Aston-Jones 2000). The present manuscript demonstrates that the threeHcrtr2 mutations known to date all result in a loss of functional receptors.
A mouse model further substantiates the link between the Hcrt/Ox system and narcolepsy. Hcrt/Ox-null mutant mice exhibit periods of behavioral arrest that strongly resemble cataplectic attacks of both humans and dogs (Chemelli et al. 1999). These mice also have a disrupted sleep architecture, particularly in the dark period, as evidenced by increased levels of both REM and NREM sleep, short latency REM periods and decreased sleep bout lengths. Along with insights from the neuroanatomy of this system mentioned above, these observations have led to the hypothesis that the Hcrt/Ox system may play a role in the normal occurrence of sleep and wakefulness. Indeed, intracerebroventricular injections of Hcrt1/Ox-A early in the light period (normally the major sleep period) increase wakefulness and reduce REM sleep in rats (Piper et al. 2000). Based on such information, we have proposed a model of arousal state control (Figure1) in which the Hcrt/Ox cells provide major excitatory input to regulate the activity of the brainstem aminergic and cholinergic areas classically implicated in the control of sleep and wakefulness (Kilduff and Peyron 2000).
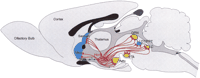
Schematic illustrating the connections from the hypocretin (Hcrt) cells (red) to some of the brain regions previously implicated in arousal state control. Aminergic nuclei are shown in gold; cholinergic regions are shown in blue. The Hcrt cells likely receive circadian input from the suprachiasmatic nuclei (SCN) shown in green, but it is not known whether such input is through a direct synaptic connection, through a multisynaptic pathway, or by a neurohumoral input. Synaptic outputs from the Hcrt cells to all regions illustrated are thought to be excitatory, based on the literature published to date. The connections between the aminergic nuclei in the brainstem and the cholinergic LDT are thought to be inhibitory. Degeneration of Hcrt neurons in human narcolepsy would result in loss of excitatory input to all these regions and, conceivably, a disruption of normal interaction between the aminergic and cholinergic cell groups. See Kilduff and Peyron (2000) for a full explanation of the model and its relevance to arousal state regulation. Based on other regulated physiological systems, it is likely that there are reciprocal connections from the brain regions illustrated to the Hcrt cells. Abbreviations: DRN, dorsal raphe nucleus; LC, locus coeruleus; LDT, laterodorsal tegmental nucleus; Nacc, nucleus accumbens; PPT, pedunculopontine nucleus; TMN, tuberomammillary nucleus; VTA, ventral tegmental area.
The Hypocretin/Orexin System and Human Narcolepsy
Although the studies cited above implicate the Hcrt/orexin system in narcoleptic dog and mouse models, the relationship between this system and narcolepsy in humans was unknown until recently. Because of the Hcrtr2 mutation in narcoleptic dogs, widespread genotyping of narcoleptic patients was undertaken. In the first published study, mutation screening of 74 patients at the Hcrt,Hcrtr1, and Hcrtr2 loci revealed a single case of aHcrt mutation in the signal peptide region which results in impaired peptide trafficking and processing (Peyron et al. 2000). This patient is among the most severely affected individuals ever observed with this disorder; symptoms were first observed at six months of age in comparison to the typical postpubertal onset. In contrast, Hcrt1/Ox-A was undetectable in cerebrospinal fluid (CSF) from seven of nine narcoleptic patients whereas it was detectable with little interindividual variation in all control subjects (Nishino et al. 2000). These results suggested a defect either in processing of the Hcrt/orexin peptides or a loss of Hcrt/orexin neurons themselves. Indeed, postmortem neuropathological studies subsequently revealed a massive reduction in the number of Hcrt/Ox neurons in the hypothalamus of narcoleptic patients (Peyron et al. 2000; Thannickal et al. 2000) with no evident change in the number of cells for melanin-concentrating hormone, a peptide coextensive with Hcrt/Ox. Thus, human narcolepsy–a disorder with unknown etiology for over a century since its original description–is likely a neurodegenerative disease.
How are these results related to the HLA association in human narcolepsy? The research focus now turns to identification of the mechanisms and cause(s) of presumptive cell death in this system, and the autoimmune hypothesis will be the immediate focus of attention. In this regard, a significant increase in the number of glial fibrillary acidic protein (GFAP)-staining astrocytes was observed in the perifornical region of the narcoleptic hypothalamus but not in the narcoleptic thalamus (Thannickal et al. 2000), consistent with a prior case report of hypothalamic gliosis in an idiopathic narcoleptic (Erlich and Itabashi 1986). Other possibilities are that an invading pathogen specifically targets the Hcrt-containing cells or that the Hcrt-containing cells are particularly sensitive to an environmental toxin.
Canine Narcolepsy Redux
Given the identification of the neuropathology of human narcolepsy as degeneration of the Hcrt/Ox cells, one might conclude that the days of the narcoleptic canine colony are numbered, particularly since narcoleptic Doberman Pinschers, Labrador Retrievers, and at least some Dachshunds have Hcrtr2 mutations rather than degeneration of the Hcrt/Ox cells. However, Hungs et al. (2001) find that, like humans, narcoleptic dogs, particularly small-breed dogs, are heterogeneous with respect to the etiology of their disorder: The novel mutation in the N-terminal region of the Hcrtr2 receptor described by Hungs et al. (2001) was found in only three of 17 cases of narcolepsy examined. Furthermore, a forthcoming paper finds that, as in humans, Hcrt1/Ox-A is undetectable in the CSF of at least two cases of sporadic canine narcolepsy but not in Hcrtr2-mutant dogs (Ripley et al. 2001). Based on the human neuropathology, one would predict degeneration of the Hcrt/Ox cells in these two cases. Given the distinct behavioral phenotype, it is likely that narcoleptic canines will remain a useful model for pharmacological studies, particularly with respect to identification of the role of the Hcrtr1 receptor.
Perspective
The good news for human narcoleptics is that no mutations have been found in either of the Hcrt receptors to date and, thus, the path to therapy is now clear: The acronym “HRT” will be redefined as “Hcrt replacement therapy” in certain circles. Although regulation of the activity of the Hcrt/orexin cells is yet to be directly addressed, given the probable role of this system in arousal state regulation and other functions such as feeding, identification of the means to manipulate the activity of these cells is likely to become an area of intense investigation. In this regard, understanding of the regulation of this gene will be of increasing interest (Waleh et al. 2001). The rapidity with which we have gone from the first description of a novel neuropeptide system with unknown function (1998), to identification of the consequence of defects in this system in animal models (1999), to understanding the neuropathology of a human disease (2000) is breathtaking and, as such, is a wonderful example of how basic research can lead to an understanding of a human disease and–hopefully soon–to new therapies.
Acknowledgments
This paper was supported by NIH grants 1 R01HL/MH59658 and 1 R01MH61755. I thank Scott Bramwell for preparation of Figure 1.
Footnotes
-
E-MAIL thomas.kilduff{at}sri.com; FAX (650) 859-5535.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.184301.
- Cold Spring Harbor Laboratory Press








