
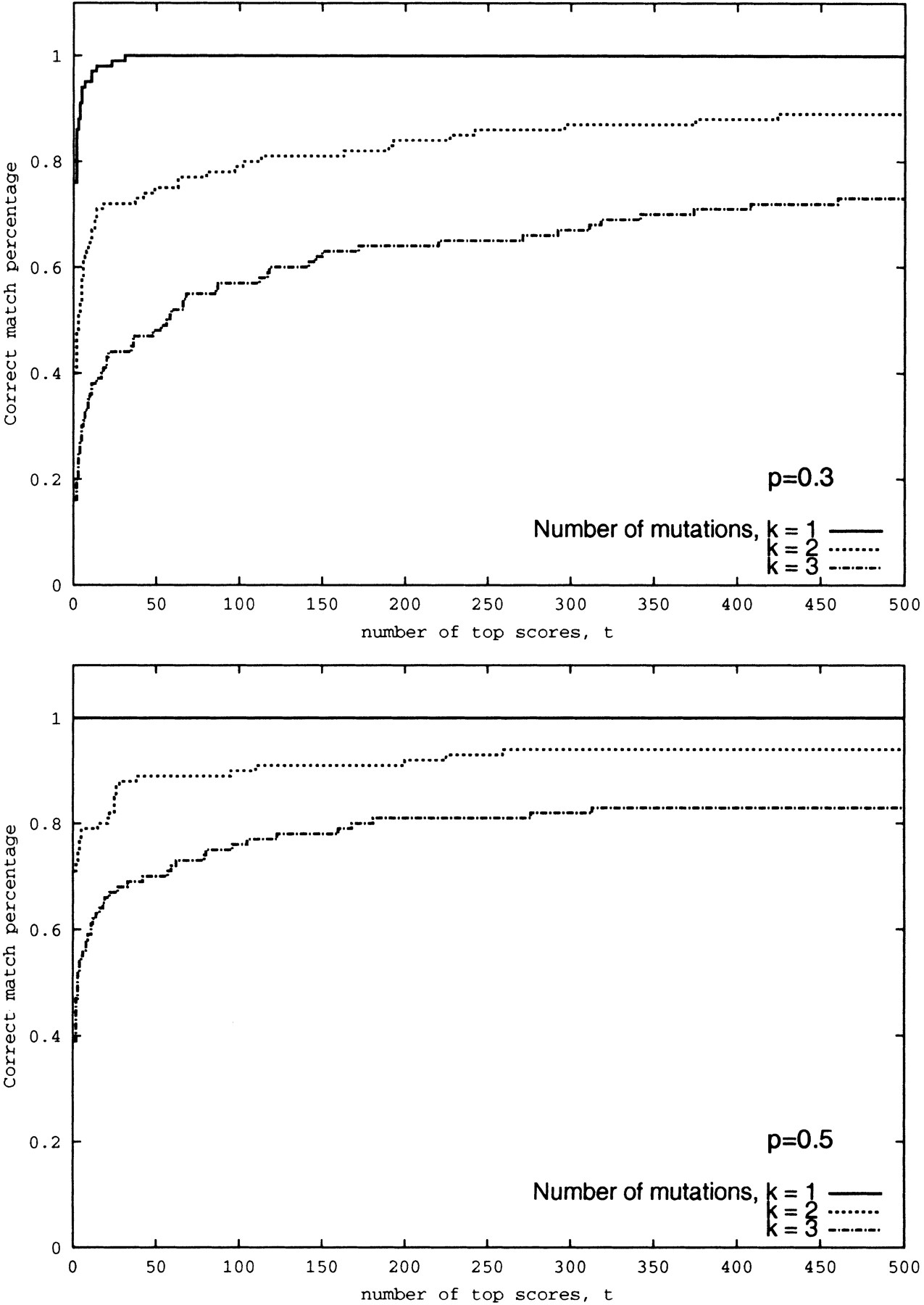
Figure 7.
Success rate of the spectral alignment approach as a function of number of top scores at qualities p = 0.3 and p = 0.5 of the simulated spectra. A match is considered correct if the correct peptide is among t top scoring peptides in the database. The database consists of 10,000 peptides.








