
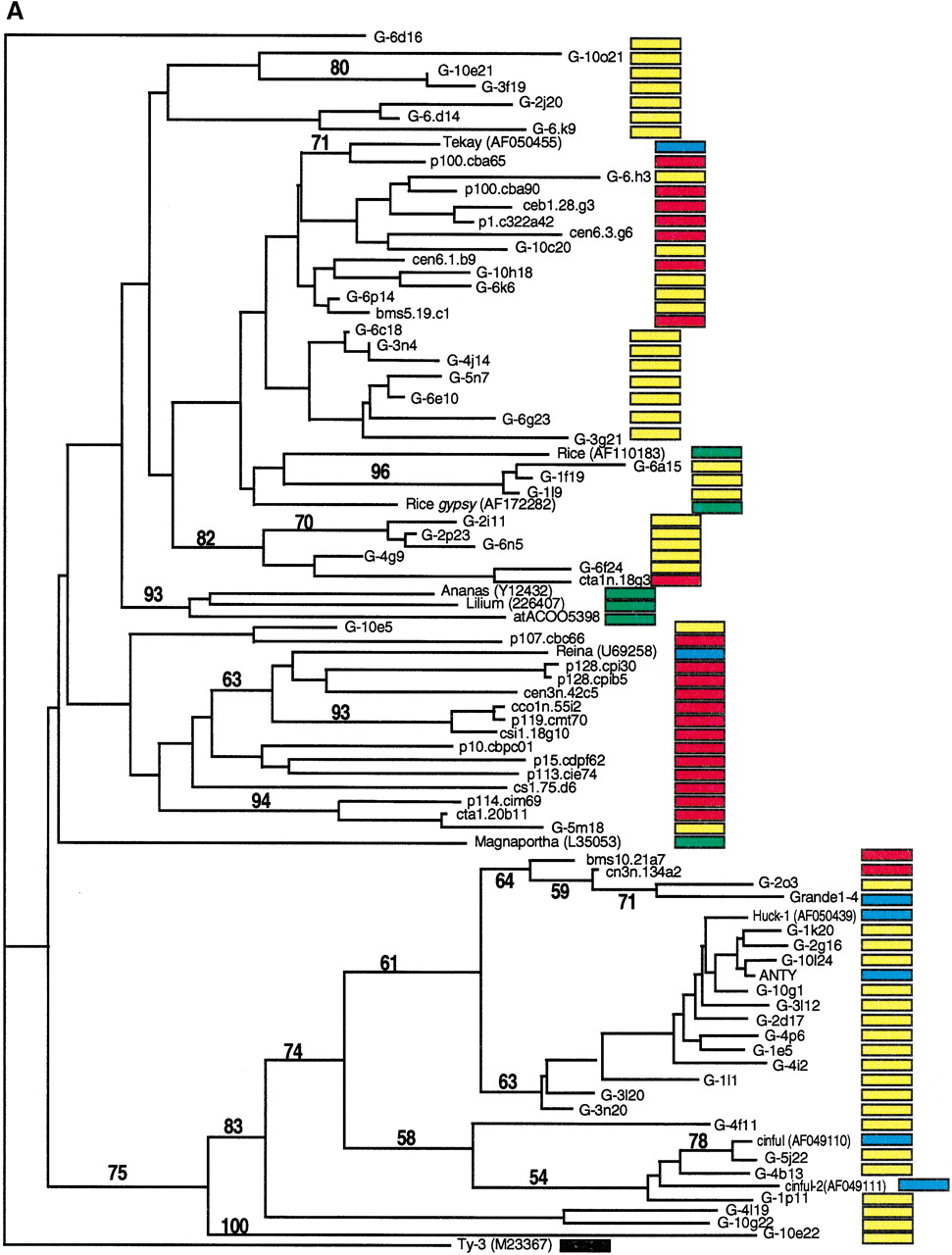

Phylogenetic analysis of LTR-retrotransposon sequences in maize ESTs and genomic sequences. All DNA sequences were translated into proteins and trimmed, and phylogenetic analyses were performed using the neighbor-joining algorithm from distance matrices according to Kimura's two-parameter method. Branch lengths are proportional to genetic distance. Bootstrap values >50 are indicated as a percentage of 1000 replicates. Maize genomic sequences from the cne1g genomic library are indicated by a yellow box to the right of the sequence; maize cDNA sequences from the DuPont database by a red box; previously described maize retroelements by a blue box; and retroelements from other species by a green box. (A) Gypsy-related sequences. Predicted proteins were homologous to a 132-amino acid region of the integrase domain. (B, next page)Copia-related sequences. Predicted proteins were homologous to a 92-amino acid region of the reverse transcriptase domain.








