
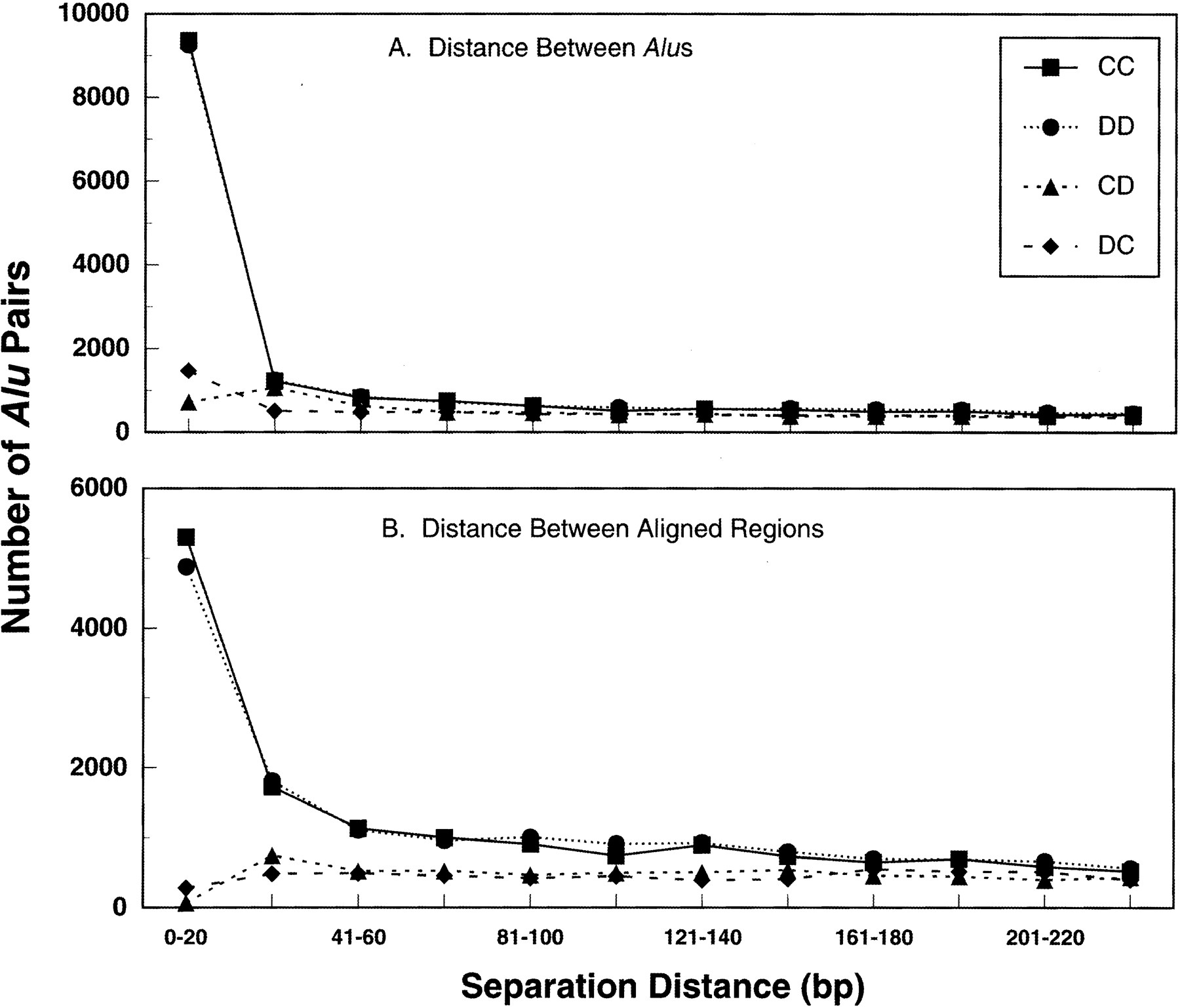
Distance between adjacent Alus versus distance between aligned regions of adjacent Alus. (A) Analysis of separation distance according to distance between Alu sequences. Nearly 30% of all direct adjacent Alus are separated by only ≤20 bp. Beyond this distance, and also for all the invertedAlus, there appears to be a random distribution of separation distances (that is, there is no preference in the distance separating adjacent Alus.).(B) Analysis of separation distance according to nucleotides separating aligned regions. Inverted repeats, especially the “tails-out” (CD) category, were uncommon. Beyond 40 bp, there appears to be a random distribution of separation distances for both the inverted and direct Alu pairs. To determine the number of occurrences of Alu pairs separated by the unique spacer distance “c”, we used the Perl program Bins C. For each of the four different orientation data sets, we ranBins using 12 different window settings between 0 and 240 bp, divided into 20-bp bins. A similar program, Bins 2, was used to subdivide the data into groups depending on the “b + c” distance, representing the distance between aligned sequences (see Methods).








