
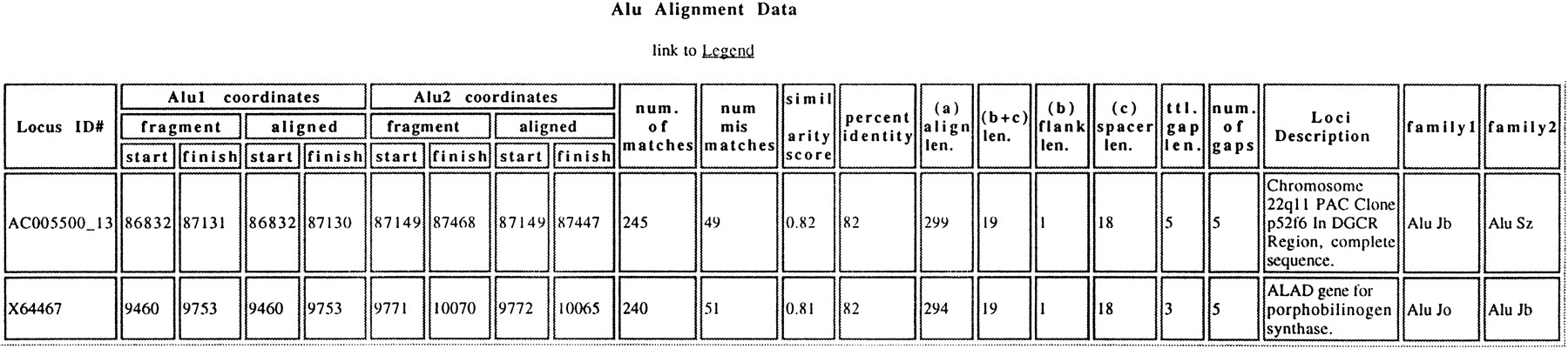
An example of an on-line summary of an adjacent pair of Alus. This table corresponds to a “cell” in the CD data table found athttp://dir.niehs.nih.gov/ALU/CD.html. It shows the only twoAlu pairs identified that meet the following criteria: the shared alignment length is between 200 and 276 bp, the pairedAlus are in the CD orientation, the separation distance b + c is between 21 and 40 bp, and there is between 86% and 90% identity. The Locus ID no., fragment coordinates, and family are extracted from the original map file. The number of matches, mismatches, similarity score, and the number of gaps are extracted from the alignment output. The locus description is extracted from the original GenBank sequence data file, as is the information necessary to determine the aligned sequence coordinates for the two fragments. Other features are described in greater detail in Methods.








