
Guilt by Association: Contextual Information in Genome Analysis
The genome sequences churned out by the “genomic revolution” have challenged both computational and experimental biologists to come up with new methods to decipher the secrets of the encoded proteins. The experimental biologists have largely concentrated on a variety of large-scale methods to assay gene expression and protein–protein interactions (Brown and Botstein 1999; Uetz et al. 2000; Walhout et al. 2000). The computational biologists, however, have deeply mined the genomes for evolutionary information in the form of homology between genes (Tatusov et al. 1997; Koonin et al. 2000;Ponting et al. 2000,). Over the past few years there has been increasing interest in the kinds of information that exist in the context in which a protein or a domain thereof is encoded in the genome (Mushegian and Koonin 1996; Dandekar et al. 1998). Recently, contextual information has been offered as a strong handle on the problem of in silico inference of protein function (Enright et al. 1999; Marcotte et al. 1999a,b; Overbeek et al. 1999; Pellegrini et al. 1999; Huynen et al. 2000). Understanding the scope and limitations of the use of these methods may be critical for the experimental biologists seeking to use computational guidelines for large-scale investigations of protein function. Here, we outline the recent advances in this direction and briefly illustrate the new leads they provide in understanding protein function.
Contextual information comes in several overlapping grades, each with a different degree of specificity with regards to a particular protein's role (Fig. 1). The most general form of contextual information is a phyletic profile, that is, the pattern of occurrence of orthologs of a particular gene in a set of genomes under comparison (Pellegrini et al. 1999; Tatusov et al. 2000). In this setup, the null hypothesis would be that genes that functionally interact in a particular pathway or complex would share a similar phyletic profile. This hypothesis is supported by the phyletic distribution of components of the core cellular machinery—the translation, transcription, and replication complexes, which interact very tightly—as well as those of metabolic pathways. For example, most of the proteins with a shared phyletic pattern between the archaea and the eukaryotes are components of one of the many protein complexes that have a role in the above-stated three-core cellular processes. Thus, the detection of uncharacterized proteins, such as the family typified by MJ0586 fromMethanococcus jannaschii, with a similar phyletic profile would implicate them in one of the core functions (Fig. 1). When the information from sequence homology is applied to these proteins, one can often arrive at rather precise functions for these proteins. In the case of the MJ0586-like proteins, sequence comparisons reveal that they have a DNA binding helix-turn-helix domain (Aravind and Koonin 1999), suggesting that it is a component of the basal transcription machinery similar to TFIIB or TBP, which share the same phyletic profile (Fig.1). This inference is compatible with the recent implication of the eukaryotic representative of this family MBF1 in transcriptional regulation (Kabe et al. 1999). When a rare shared phyletic pattern is seen for certain proteins whose sequence affinities suggest a related function, a strong case can be made for their interaction. One example of this is the typically eukaryotic chromatin protein methyltransferase—the SET domain that is seen in the bacteria Chlamydiae and Bordatella pertussis, along with another eukaryotic chromatin protein domain, SWIB (Stephens et al. 1998). The rare phyletic profile of these proteins in bacteria suggests that the SET and SWIB domains probably interact not only in these bacteria but possibly also in other organisms.
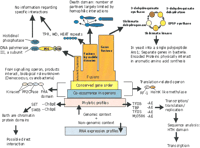
A schematic representation of the different grades of contextual information in the rough hierarchy of their specificity. It should be kept in mind that the grades overlap to some extent. The different specific examples that have been discussed in the text are illustrated for each grade. The simplified phylogenetic profiles follow. -AE stands for present only in archaea and eukaryotes, while -ChBpE stands for present in Chlamydiae, Bordetella, and Eukaryotes but absent in other prokaryotes. PHP is the phosphoesterase domain found in the DNA polymerase III subunits and histidinol phosphatases and the H for the HhH DNA binding module. In the case of the Death domain, the potential interactions between Death domains in different proteins present in a given genome are shown. The enzymes of the aromatic amino acid biosynthesis pathway that are encoded by separate genes in bacteria but fused into a single polypeptide in yeast are shown as a case of gene fusion.
Since the pioneering works of Jacob and Monod, scientists have realized that functionally linked genes are coregulated and occur in proximity to each other on the chromosome. Genome comparisons have supported this and show that, in prokaryotes, functionally interacting genes are in clusters that range from the giant ribosomal operons to gene pairs that survive over large phylogenetic distances (Dandekar et al. 1998;Overbeek et al. 1999). Thus, the occurrence of an uncharacterized gene in the neighborhood (the same operon) of genes with known functions could potentially betray its function. However, the variability of operons and the inability to predict them with a high level of certainty causes a loss of specificity of this form of contextual information. On this issue, Huynen et al. argue that greater stringency in the criteria for gene neighborhood reduces false positives in these inferences, with physical interactions between gene products strongly predicted by conservation of gene order in an operon. Furthermore, as the number of gene neighborhood combinations are far from exhausted with the currently available genomes, this method is likely to improve in its scope and confidence with the availability of more genomes in the future. While establishing functional interactions between the proteins, the operon conservation approach has a deep predictive value only for protein complexes that function in metabolism and core processes. This is mainly because the functions of the uncharacterized proteins can be interpreted in light of a fairly well-known biochemical pathway. There are several conserved operons that encode proteins such as histidine kinases and chemotactic receptors and ς regulatory kinases and phosphatases that are likely to physically interact in distinct signaling pathways. Contrary to the metabolic- and core-function proteins, in most such cases no specific biological role can be assigned for these predicted signaling pathways (Fig. 1).
Protein domain fusions provide another form of information that could help in unraveling uncharacterized proteins or domains. Here the ability to extract useful information is very contingent on the proper classification of domain fusions. In evolutionary terms domain fusions can be classified as follows.
First, there are the ancient fusions that have occurred in the common ancestor of a major lineage of organisms, such as the domains comprising the α-subunit of the bacterial DNA polymerase III. In this case the protein is made up of a phosphoesterase domain, a DNA polymerase region, and two DNA-binding modules—a Helix-hairpin-Helix (HhH) and an OB domain (Fig. 1). These domains by themselves provide information only in the context of the DNA polymerase, rather than automatically extending to the other proteins in which they are found, such as the histidinol phosphatase that also has the same phosphoesterase domain. Thus, they are of limited value in predicting potential interaction partners of these proteins.
Second are the recent fusions that have occurred in the more terminal branches of the tree of life (though they may show a secondary dispersion through horizontal transfer). The second class of recent fusions may further be divided into those driven by evolutionarily mobile domains (‘promiscuous domains’; Marcotte et al. 1999a) and gene fusions that arise from fusions of genes that typically occur separately in several other genomes. A good number of the commonly encountered mobile domains, which include repetitive modules such as β-propellers and α-α superhelices, function in numerous distinct protein–protein interactions in different cellular compartments and are of minimal value for predicting specific interactions. Other fusions between mobile domains such as helicases and nucleases illustrate a general tendency for these domains to physically interact (Aravind et al. 1999), but this does not imply that all related nucleases and helicases interact. Some of the mobile domains such as the Death domains in animal apoptotic signaling mediate homophilic interaction, while cysteine knot domains often interact with extracellular leucine-rich repeat domains. Hence, the presence of such domains in proteins can often narrow down, if not specify, the set of interaction partners in a given genome. Other mobile domains such as the HhH module or the ACT domain act as tags specifying interactions with nucleic acids and small molecules, respectively, and predict the protein's functional milieu to some extent. Unlike the wide spectrum of relatively imprecise predictive information offered by the mobile domain fusions, the relatively recent gene fusions are generally direct indicators of physical interactions between the proteins encoded by separate versions of these genes (Marcotte et al. 1999a). Such fusions are typical in proteins that are parts of the same complex or catalysts of adjacent reactions in pathways (Fig. 1) and that are, in prokaryotes, extreme cases of gene order conservation in operons. While this form of fusion has a high predictive value distinguishing it from other the forms, fusions may not necessarily be straightforward in the absence of a robust list of orthologs (Galperin and Koonin 2000).
What is the total amount of contextual information with all its overlapping grades that is available for a particular genome? Huynen et al. (2000) determine this for the smallest cellular genome available to us, that of Mycoplasma genitalium. They show that such information can be gleaned for up to 50% of the genes with gene order conservation contributing the maximum information and gene fusions, phyletic profiling, and co-occurrence in operons contributing much smaller, roughly equivalent amounts of information. The observation that gene order conservation provides most of the contextual information has serious implications for the generality of these methods because the operonic organization of genes is a hallmark of the prokaryotes. A large part of the critical regulatory interactions of eukaryotic proteins, especially typical of multicellular forms, cannot avail itself of this form of contextual information. A partial solution to this problem is the use of coexpression, as determined through transcription profiling, as an additional form of nongenomic contextual information in the eukaryotes (Marcotte et al. 1999b). While expression profiles are far from being comparable to operons in their predictive specificity, multiple combined approaches that additionally use the results of large-scale genetic screens and mass spectroscopy–based characterization of protein complexes could increase the amount of context information in eukaryotic systems.
Finally, it can be seen that leads into fundamental biological processes such as translation may be derived from contextual methods if they are suitably combined with robust homology-based characterization of the concerned proteins. Different forms of contextual information have pointed out the role of at least two GTPases, one of the TdhF family (Huynen et al. 2000) and another of the OBG1 family fused with a predicted RNA binding TGS domain (Galperin and Koonin 2000). They are likely to participate in translation, suggesting as yet undiscovered GTP-dependent steps. Using similar techniques, different enzymes, namely at least two methyltransferases (MG259 and MG347; Huynen et al. 2000) and one Rossmann fold oxidoreductase of the GidA family (Marcotte et al. 1999b), have been implicated in translation-related functions in bacteria and mitochondria. Both MG259 and MG347 are similar to RNA methylases, and least one of them (MG259) has signatures typical of nucleic acid adenine methylases. This, taken together with the link to translation, implies that they are involved in methylation (MG259 and MG347) and base modification (GidA) of either tRNA or rRNA. Thus, in conjunction with appropriate homology studies, ‘guilt by association’ can be converted to clear-cut hypotheses that are testable with specific experiments.
Experimental methods such as high-throughput two-hybrid screens (Uetz et al. 2000; Walhout et al. 2000) suffer from false positives arising because of interactions among proteins from different subcellular locations. In such cases, the contextual information could effectively cut down the search space for interacting partners and thereby improve the chances of finding biologically relevant interactions. With genome sequence data pouring in, these computational approaches are likely to go hand in hand with large-scale experimental efforts in the conquest of unchartered biological systems.








