
Comparative Genomics Sheds Light on Mechanisms of Genomic Imprinting
Genomic imprinting is an epigenetic chromosomal modification in the germ line that leads to preferential expression of one of the two parental alleles in a parent-of-origin-specific manner. A number of recent studies suggest that genomic imprinting is mediated by a set of elements in distinct chromosomal regions that have been termed imprinting centers (IC). By definition, the IC coordinate three activities: the establishment of imprint marks, the maintenance of these imprint marks throughout development, and the implementation of the preferential expression from a specific parental allele (for review, see Ben-Porath and Cedar 2000).
At least four major imprinting models that incorporate recent discoveries have been elucidated: First, the sense/antisense competition model for preferential allelic expression, as exemplified by studies examining the regulation of Igf2r and its associated antisense on mouse chromosome 17 (Box ; Fig.1). Second, the enhancer/chromosomal insulation model developed on the basis of the reciprocal imprinting of Igf2/H19 in human and in mouse (Box ; Fig.2A). Third, the bipartite IC model, which describes the regulation of multiple imprinted genes in a 2-Mb Prader-Willi syndrome/Angelman syndrome(PWS–AS) region of human chromosome 15. Finally, the promoter-specific reciprocal-imprinting model as seen at the human and mouse Gnas locus (Box ; Fig. 2B).
Imprinting of Igf2r: Sense and Antisense Competition Model (Fig. 1)
An elegant model of sense/antisense competition has been proposed by Wutz et al. (1997). On the mouse paternal allele, the IC in region 2 is unmethylated, while promoter region 1 is methylated; antisense Air is transcribed. Aircompetes and turns off the Igf2r sense transcript. On the maternal allele, the IC is methylated and Air is turned off. Promoter region 1 is unmethylated, and Igf2r is transcribed. Two cis-regulatory elements in region 2, the de novo methylation signal (DNS) and the allele-discrimination signal (ADS), have been shown to be involved in the establishment of differential methylation of the Igf2r DMR (Birger et al. 1999). Imprinting of Igf2r is tissue specific. In the mouse central nervous system (CNS), Igf2r is transcribed from both parental alleles (Wang et al. 1994). On the paternal allele, the IC is unmethylated, and Air is transcribed. The promoter region 1 is unmethylated; Air transcription fails to outcompete Igf2r on the paternal allele (Hu et al. 1999). Imprinting of Igf2r is species specific. In humans, the “homologous IC” is methylated on the maternal allele. On the paternal allele, unmethylated IC fails to initiate antisense transcript (T.H. Vu and A.R. Hoffman, unpubl.). The human homologous IC is inactive. Promoter region 1 is unmethylated on both alleles;IGF2R is not imprinted. We propose that the active IC in the mouse coordinates the correct methylation pattern in promoter region 1 and initiates antisense transcription. In mouse CNS, IC fails to establish the correct promoter methylation, while in humans the “defunct IC” fails in both activities. Imprinting ofIgf2r is also observed in marsupials (mammals that give birth to altricial offspring) but this imprinting is not observed in monotremes (egg-laying mammals) (John et al. 2000; Killian et al. 2000). However, no differential methylation is detected in region 2 of the marsupial Igf2r. The mechanism of marsupialIgf2r imprinting remains to be explored. Since the discovery of Air, imprinted antisense transcripts are observed in all four major imprinting models (see text). In humanKVLQT1, targeted deletion of the LIT1 CpG island (the putative IC) abolishes transcription of the antisense on the paternal allele, accompanied by activation of multiple normally suppressed paternal alleles in the KVLTQ region (Horike et al. 2000).
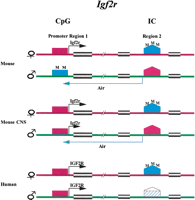
Coordination of IC methylation, promoter methylation, and antisense transcription in the Igf2r imprinting model. Red box, unmethylated and active promoter. Green box, methylated and inactive promoter. Red pentagon, unmethylated and active IC. Green pentagon, methylated IC. White pentagon, unmethylated and defunct IC. Black arrow, sense transcript. Purple arrow, antisense transcript.
Imprinting of Igf2/H19 andGnas: Reciprocal Imprinting and Chromosomal Insulation Model (Fig. 2)
Reciprocal imprinting of Igf2/H19 has been studied extensively. The imprinting of human DLK1/GTL2(mouse Dlk1/Gtl2) is similar to Igf2/H19imprinting. On the maternal allele, the IC is unmethylated. CTCF binds to the IC, which insulates enhancer activity from Igf2,only H19 is transcribed. On paternal allele, the IC is methylated, which prevents CTCF binding. The IC also spreads CpG methylation along the H19 promoter. Only Igf2is transcribed (sense and antisense). The Ihit1 transcript is located 20 kb upstream of H19 (Onyango et al. 2000). The imprinting status of Ihit1 has not been determined. Reciprocal imprinting in Gnas is more complex.Nesp and Xlas transcripts are reciprocally imprinted, while Gsα is transcribed from both parental alleles (Liu et al. 2000). A putative IC, 2 kb upstream ofGsα, is methylated on the maternal allele, which may act on the methylation of the maternal Xlas promoter.Nesp is transcribed from the unmethylated maternal promoter. On the paternal allele, the IC is unmethylated and so is theXlas promoter. Both Xlas and the IC (exon 1A) are transcribed (Liu et al. 2000). Antisense (Gnas-AS orNesp-AS) is also transcribed from the unmethylatedXlas promoter region (Li et al. 2000; Wroe et al. 2000), while the methylated Nesp promoter turns offNesp transcription. Enhancer activity and a chromosomal boundary have not yet been found.
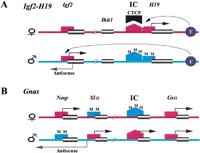
(A) Coordination of IC methylation, promoter methylation, and enhancer blocking activity in the Igf2/H19 reciprocal imprinting model. CTCF, enhancer-blocking protein. Ihit1,novel Igf2-H19 interval transcript. (B) Coordination of IC methylation, promoter methylation, and antisense transcription in Nesp/Xlas/exon 1A reciprocal imprinting model.
To understand the mechanisms underlying genomic imprinting, a number of research groups have focused on the identification and characterization of the imprinting elements present in the various ICs. As the DNA sequence of the whole human genome is nearly complete and complete sequences of other species are or will soon be available, an entirely new strategy for understanding potential imprinting mechanisms has now become available. Sitting at one's computer, it will be possible to devise large-scale comparisons of genomic sequences within and/or between species that could yield new information on potentially important elements that govern preferential allelic expression.
In this issue, we see how two research groups (Wylie et al. 2000;Onyango et al. 2000) have advanced our understanding of the mechanisms of genomic imprinting by employing a comparative analysis of distinct imprinting regions in human and mouse genes.
Human Chromosome Regions Have Common Imprinting Elements
R. Jirtle and his colleagues at Duke University have identified two reciprocally imprinted genes, DLK1 and GTL2, on human chromosome 14q32 (Wylie et al. 2000). The cognate murine genes,Dlk1 and Gtl1, on mouse chromosome 12 are also linked and reciprocally imprinted (Schmidt et al. 2000; Takada et al. 2000).DLK1, which encodes a transmembrane protein with 6 EGF (epidermal growth factor) repeat motifs, is paternally expressed. In contrast, GTL2, which encodes an apparently nontranslated RNA, is located 102 kb downstream of DLK1 and is expressed from the maternal allele.
A functionally similar imprinted region that harbors two reciprocally imprinted genes, IGF2 and H19, has been extensively studied in both mouse and human (Box ). There are striking similarities in the spatial, structural, and reciprocal imprinting characteristics of these two imprinted regions (IGF2/H19 on human chromosome 11p15 andDLK1/GTL2 on human chromosome 14q32). IGF2is the paternally expressed gene, while the maternally expressedH19 encodes for a nontranslated RNA. These similarities have prompted the Duke group to further elucidate the IC elements on 14q32 region by making careful comparisons with the 11p15-imprinted region.
CTCF is a zinc finger protein implicated in the regulation of chromosomal boundaries (Fig. 2A). In the mouseIgf2/H19 region, CpG methylation in the CTCF binding site eliminates binding of the enhancer-blocking CTCF, thereby allowing expression of Igf2 from the paternal allele (Bell and Felsenfeld 2000; Hark et al. 2000). Methylation analysis by bisulfite sequencing revealed a differential methylation region (DMR) within 2 kb upstream of GTL2, analogous to the silencer DMR1 of the IC on the IGF2/H19 (Wylie et al. 2000). A DMR in the promoter region of DLK1 may correspond to the DMR in theIGF2 gene (Constancia et al. 2000). Interestingly, two unique CTCF binding sites were found in the region one kilobase upstream ofGTL2. Bisulfite sequencing indicated that all CpG sites within and outside the binding sites were hemimethylated (equal numbers of methylated and unmethylated molecules), suggesting that CpG methylation in this DMR may serve as an epigenetic switch to turn on/off the enhancer-blocking activity of the IC by binding/unbinding the CTCF to the unmethylated/methylated IC. In parallel with the enhancer/chromosomal-boundary model of reciprocal imprinting, Wylie et al. (2000) also found the same consensus sequence for two enhancer elements as seen in IGF2/H19 ∼10 kb downstream from the GTL2 transcription site.
Wylie et al. (2000) have presented an elegant approach combining bioinformatics and experimental data on allelic expression and bisulfite sequencing to elucidate a model forDLK1/GTL2 reciprocal imprinting. TheDLK1/GTL2 and the IGF2/H19 regions are located on different chromosomes (human 14q32 and 11p15; mouse chromosomes 12 and 7), and they encode functionally diverse proteins DLK-1 (or FA1) and IGF-II. The striking similarities in the imprinting characteristics of these two imprinting dyads suggest that many imprinting elements that are crucial for this type of reciprocal imprinting would be conserved in heterologous chromosomal locations within one species, as well as in orthologous regions between species.
The Structure of a 1-Mb Imprinted Region
Imprinted genes usually appear in pairs or clusters. As imprinting often influences large chromosomal domains and may be regulated by more than one (or by a bipartite) IC, comparison of larger imprinted domains would pinpoint the crucial conserved, putative imprinting elements in the ICs. In this issue, A. Feinberg (2000) and his colleagues at Johns Hopkins University pioneered such an approach (Onyango et al. 2000). They performed a comparative analysis of a mouse 1-Mb imprinted region that is orthologous to the human 11p15 domain. They constructed a BAC contig containing ∼1 Mb of the imprinted region on mouse chromosome 7. By comparing this region with its orthologous region on human chromosome 11p15, they were able to identify 33 conserved CpG islands (with sequence identity ≥50%) and 49 conserved nonexonic, non-CpG island sequences (at least 70% sequence identity in 100 bp). This group and others (Smilinich et al. 1999; Paulsen et al. 2000) have previously shown that the human imprinted region harbors two imprinted subdomains: the IGF2/H19 domain and the domain centered around KvLQT1. These domains are separated by a nonimprinted domain containing TSSC6-TAPA1-TSSC4-TRPC51. Similar findings pertain to the mouse.
All of the imprinted genes in the mouse contain at least two CpG islands, except for the antisense Lit1 transcript, and they are clustered by intronic sequence elements that are conserved compared with the human gene cluster. By doing this comparison, Onyango et al. (2000) identified two transcripts unique to mouse and five transcripts unique to human. In the Kvlqt1 domain, a conserved novel transcript, Tssc8, was identified; it lies within both theKvlqt1 and antisense Lit1 transcripts. Tssc8has no obvious ORF and is transcribed in the opposite orientation fromLit1, suggesting that it represents a novel antisense of an antisense (Lit1) transcript. The imprinting status and potential function of Tssc8 will require further investigation. In addition, a mouse-specific paternally imprinted transcript (Msuit) was discovered within the Kvlqt1domain upstream of p57kip2.
Within the Igf2/H19 imprinted domain, they identified a novel conserved transcript 20 kb downstream of H19 and a novel Igf2-H19 interval transcript (calledIhit1) 20 kb upstream of H19 (Fig. 2A). Determination of the imprinting status of the novel downstream transcript will help elucidate the boundary of the imprinted/nonimprinted subdomains. The imprinting status of the interval transcript Ihit1 will provide further evidence to test the enhancer/chromosome boundary model in the Igf2/H19 imprinted domain (Box ; Fig. 2A).
Future Developments
Sequence comparisons of large DNA regions across species have been used recently to identify long-range regulatory elements that are difficult to discover by conventional methods. A coordinate regulator of interleukins 4, 13, and 5 was identified by large-scale sequence comparison of ∼1-Mb human 5q31 region and the murine chromosome 11 orthologous region (Loots et al. 2000). By comparing imprinted domains in the mouse to the orthologous human sequences, Onyango et al. (2000)have identified multiple putative imprinting elements in theIgf2/H19 and Kvlqt1 imprinted domains. The full characterization of these putative imprinting elements will undoubtedly be forthcoming. Further comparisons of orthologous sequences in other mammals may help to pinpoint crucial imprinting elements, as demonstrated by the recent innovative studies that have elucidated the evolution of Igf2r imprinting (John et al. 2000; Killian et al. 2000).
An alternative yet complementary approach is to compare sequences of similar imprinted domains located in different loci but within the same genome, as reported by Wylie et al. (2000). In the next few years, large-scale sequence comparisons of various imprinted regions within one species and sequence comparisons across species are certain to provide important new insights on various imprinting elements of the IC.
The IC implements preferential expression of a specific parental allele through imprint marks that are established in the germ line during gametogenesis and are maintained throughout development. Although the gametic imprints can be modulated by differential chromatin structure and/or by differential DNA replication timing, the primary imprints often involve DNA methylation.
Methylation of cytosine in CpG dinucleotides creates the fifth base, a methylated cytosine, along with the four conventional DNA-building blocks, G, A, T, and C. Bisulfite sequencing, as used by Wylie et al. (2000) and others (e.g., Vu et al. 2000), helps to elucidate the imprint marks of the methylated CpG in the IC.
The methylation of a CpG island in the promoter region of a gene is a hallmark of gene regulation. The notion by Onyango et al. (2000) that almost all imprinted genes are associated with two or more CpG islands suggests the complexity of the imprinting mechanisms. Gametic methylation of IC elements located far away from the promoter region can induce methylation/demethylation of the promoter CpG islands, thus allowing “working from a distance.” IC methylation acting incis and trans can coordinate sense/antisense transcripts through direct promoter methylation, chromatin remodeling (e.g., by recruiting histone deacetylase), or acting on a chromosomal boundary with enhancer-blocking activity.
DNA methylation plays a pivotal role in genomic imprinting. Further innovative developments in quantifying CpG methylation genome wide (such as the development of CpG DNA chips) or quantifying methylation at specific sites (such as methylation restriction [MR], MR-PCR; T.H. Vu and A.R. Hoffman, unpubl.) would be of great interest.
[For more information on comparative genome analysis and imprinting, see Okamura et al. in the December issue of Genome Research.]
Acknowledgments
We thank all members of our laboratory for technical support, especially Tao Li for designing the figures. This paper was supported by NIH grant DK36054 and the Research Service of the Department of Veterans Affairs.
Footnotes
-
↵1 Corresponding authors.
-
E-MAIL thanvu{at}stanford.edu, arhoffman{at}leland.stanford.edu; FAX (650) 856-8024.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.166200.
- Cold Spring Harbor Laboratory Press








