Assembly properties for assemblies with simulated reads of Saccharomyces cerevisiae chromosome 5 (coverage 130-, 260-, 138-fold) and chromosome 7 (coverage 270-, 135-, 146-fold) and the complete genomes of Haemophilus influenzae (coverage 154- and 164-fold) and Escherichia coli (coverage 95-, 127-, 135-fold)
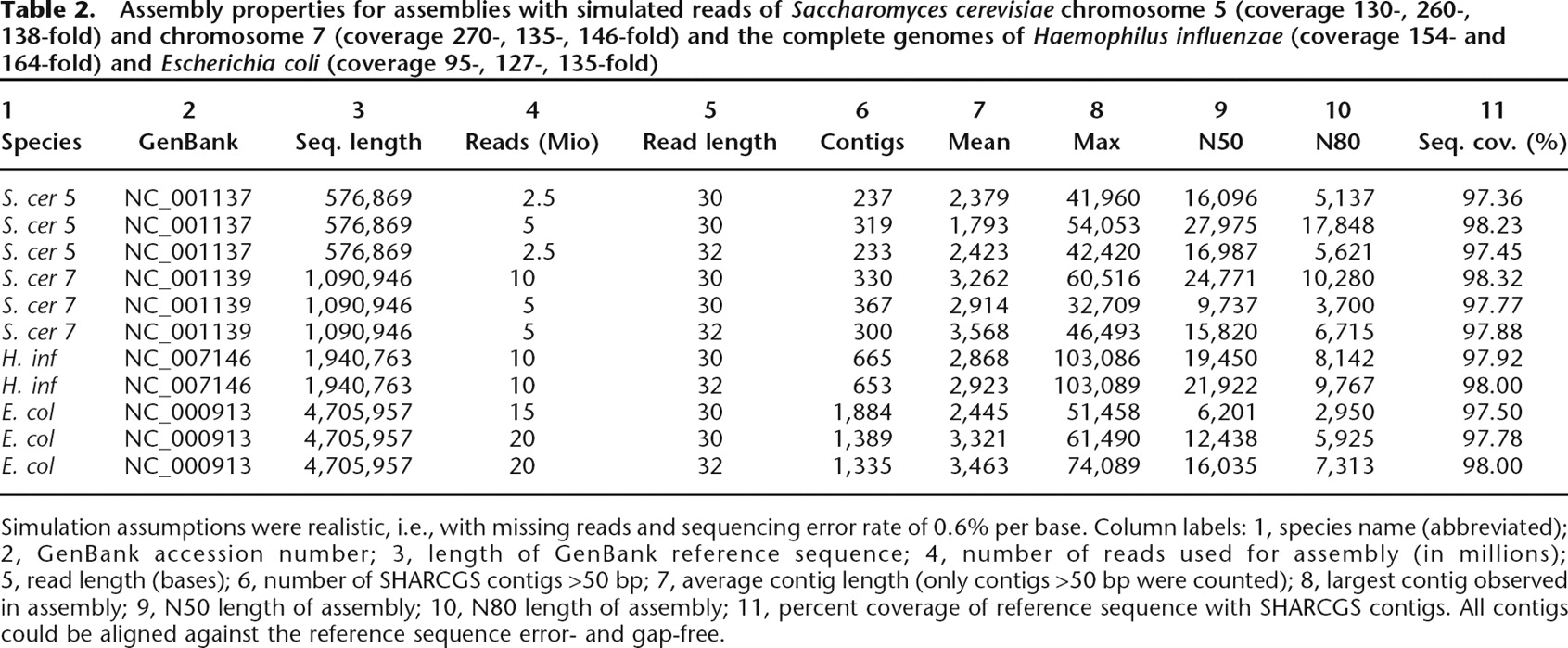
Simulation assumptions were realistic, i.e., with missing reads and sequencing error rate of 0.6% per base. Column labels: 1, species name (abbreviated); 2, GenBank accession number; 3, length of GenBank reference sequence; 4, number of reads used for assembly (in millions); 5, read length (bases); 6, number of SHARCGS contigs >50 bp; 7, average contig length (only contigs >50 bp were counted); 8, largest contig observed in assembly; 9, N50 length of assembly; 10, N80 length of assembly; 11, percent coverage of reference sequence with SHARCGS contigs. All contigs could be aligned against the reference sequence error- and gap-free.