Comparative analyses of the genomes and proteomes of five teleost fishes
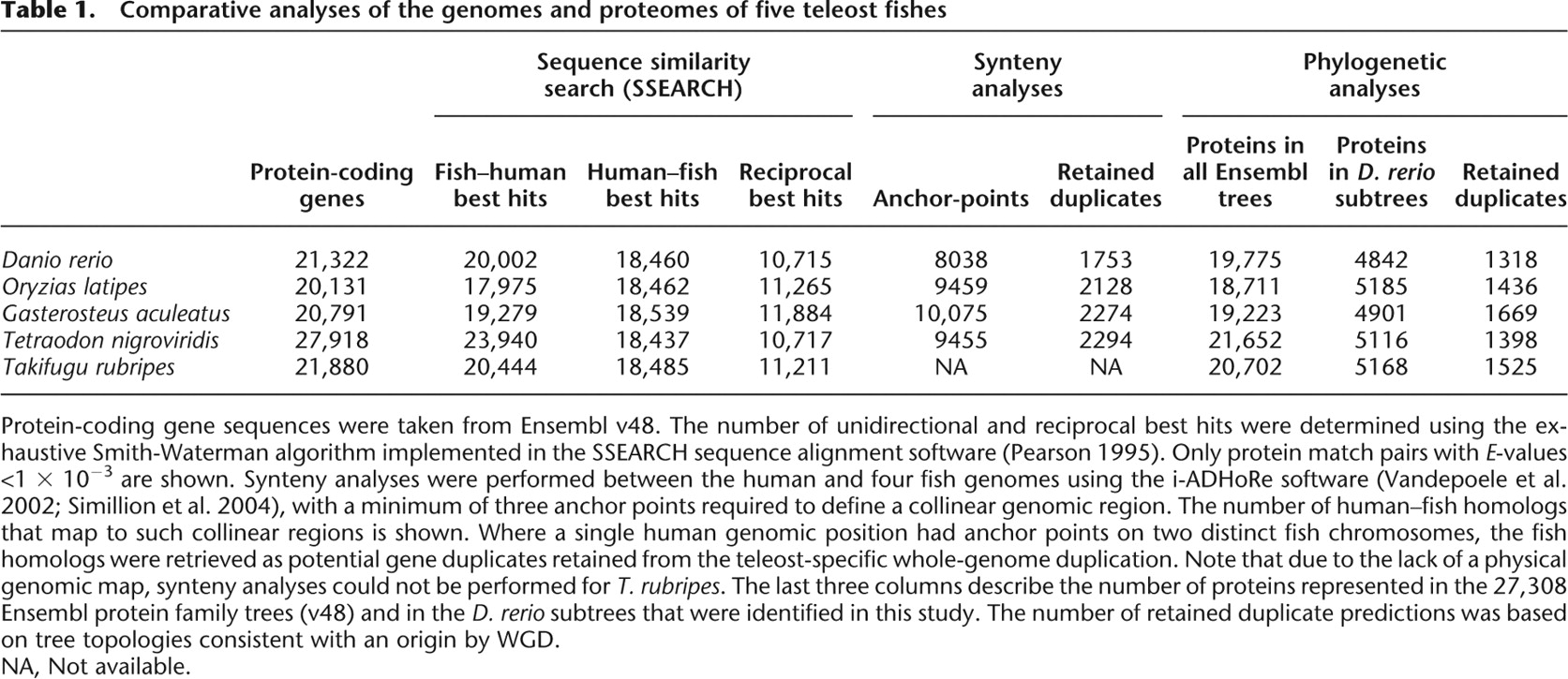
[i] Protein-coding gene sequences were taken from Ensembl v48. The number of unidirectional and reciprocal best hits were determined using the exhaustive Smith-Waterman algorithm implemented in the SSEARCH sequence alignment software (Pearson 1995). Only protein match pairs with E-values <1 × 10−3 are shown. Synteny analyses were performed between the human and four fish genomes using the i-ADHoRe software (Vandepoele et al. 2002; Simillion et al. 2004), with a minimum of three anchor points required to define a collinear genomic region. The number of human–fish homologs that map to such collinear regions is shown. Where a single human genomic position had anchor points on two distinct fish chromosomes, the fish homologs were retrieved as potential gene duplicates retained from the teleost-specific whole-genome duplication. Note that due to the lack of a physical genomic map, synteny analyses could not be performed for T. rubripes. The last three columns describe the number of proteins represented in the 27,308 Ensembl protein family trees (v48) and in the D. rerio subtrees that were identified in this study. The number of retained duplicate predictions was based on tree topologies consistent with an origin by WGD.
[ii] NA, Not available.