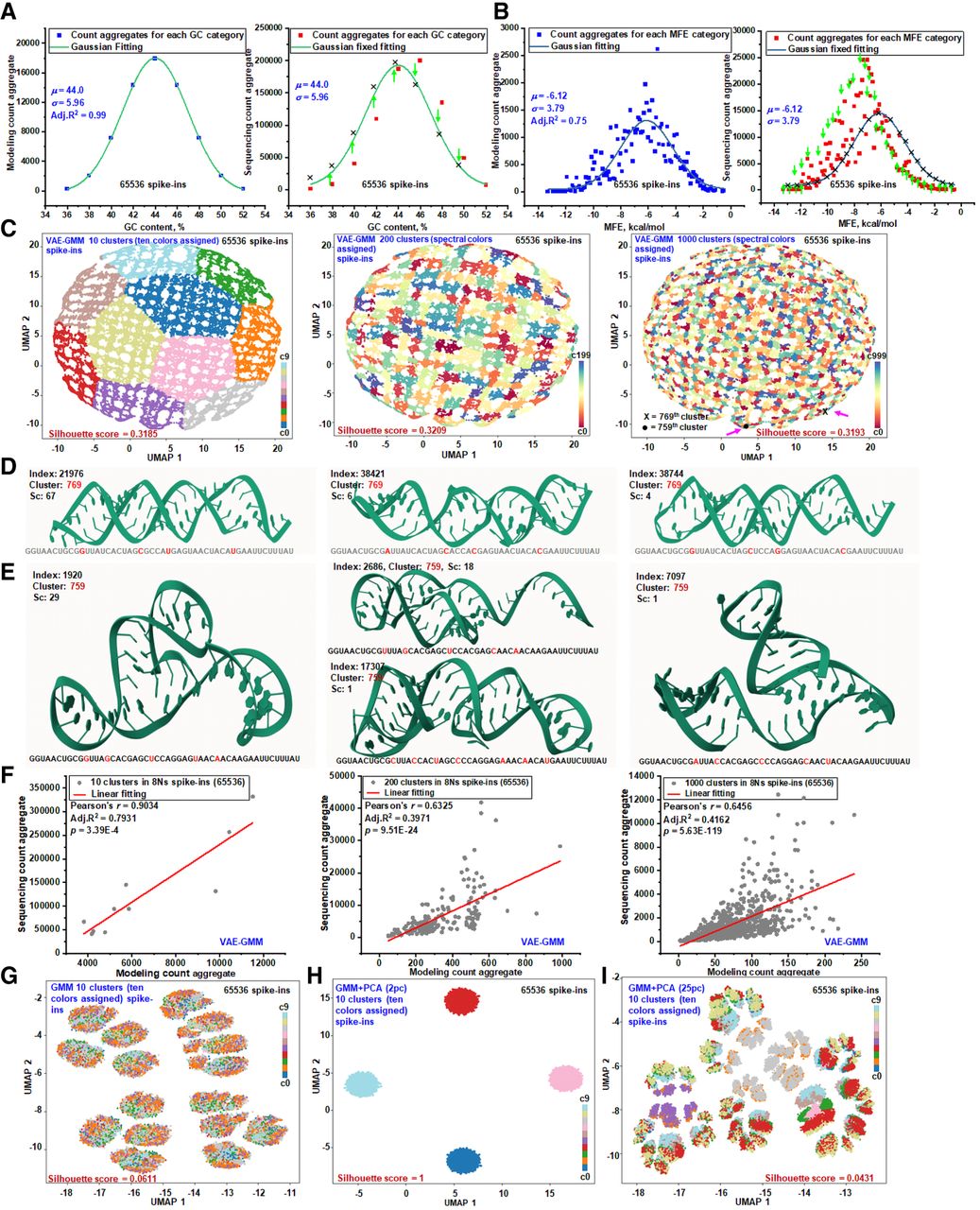
RNA structure–driven sequencing biases from primary to tertiary level. (A) Gaussian modeling of spike-in RNAs by GC-content. A total of 65,536 spike-in RNAs were categorized based on their GC-content to establish calibration benchmarks assuming a uniform distribution. Sequencing data are organized by GC-content and aligned using parameters derived from the Gaussian model. Arrows indicate discrepancies between the model's predicted counts and the actual sequencing data at various GC-content levels. (B) Gaussian modeling of spike-in RNAs by MFE. The same set of spike-in RNAs was categorized by MFE values (binned to one decimal place) to set up calibration benchmarks under a uniform distribution assumption. Sequencing data are organized by MFE and adjusted to align with the Gaussian model's parameters. Arrows highlight variances between the predicted counts and the actual sequencing data at different MFE levels. (C) UMAP visualization of VAE-GMM clustering at multiple resolutions. UMAP plots display the clustering results of the spike-in RNAs using a VAE-GMM at three levels of granularity: 10, 200, and 1000 clusters. Each plot shows the distribution of spike-ins across the identified clusters. Clustering performance at each level is quantified using the silhouette score, indicating the degree of cluster separation. (D,E) AlphaFold-predicted 3D structures from distinct VAE-GMM clusters. Three-dimensional structures predicted by AlphaFold are presented for selected spike-in RNAs from two distinct clusters highlighted in panel C at the 1000-cluster level. For each spike-in, the RNA index, cluster ID, nucleotide sequence, and sequencing count (sc) are provided. These structures illustrate the diversity of RNA folding within different clusters. Mol* is used for 3D structure visualization (Sehnal et al. 2021). (F) Aggregated modeling and sequencing counts across VAE-GMM clusters. The modeling predictions and actual sequencing counts are aggregated across VAE-GMM clusters at multiple scales (10, 200, 1000 clusters). This comparison illustrates how complex RNA structural features influence local sequencing efficiency, as reflected in discrepancies between predicted and observed counts. (G) UMAP visualization of GMM clustering on one-hot encoded sequences. Spike-in RNAs are clustered using GMM based on one-hot encoded nucleotide sequences into ten clusters. The UMAP plot visualizes the distribution of RNAs across these clusters, with silhouette scores assessing the quality of cluster separation. (H) GMM clustering with PCA reduction to two components. The spike-in RNAs are clustered using GMM after reducing the data to two principal components via PCA, presetting the number of clusters to 10. The resulting UMAP plot shows the spatial arrangement of clusters in reduced dimensions. (I) GMM clustering with PCA reduction to 25 components. Similar to panel H, but PCA reduces the data to 25 principal components before GMM clustering into 10 clusters. Silhouette scores are presented to evaluate cluster separation. In all UMAP visualizations, clusters are color-coded to represent different groups.