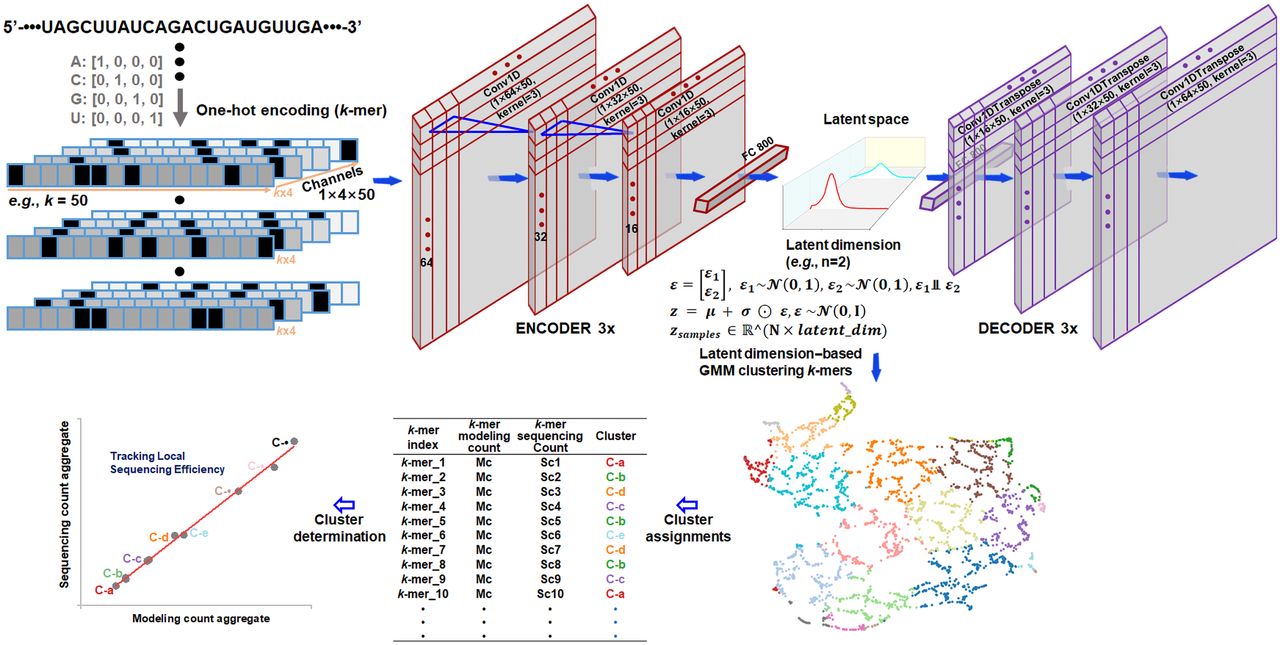
Architecture and training pipeline of the VAE-GMM k-mer clustering model for examining high-dimensional structures unveiling bias mechanisms in RNA-seq. The k-mer RNA sequences are one-hot encoded and processed through an encoder consisting of three one-dimensional CNN layers and a dense layer that outputs mean (µ) and standard deviation (σ) vectors for the VAE's latent space. Latent variables z are sampled using the reparameterization trick z = µ + σ ⊙ ε, where . The decoder mirrors the encoder architecture. The VAE is trained by optimizing a loss function that combines reconstruction loss and Kullback-Leibler divergence. After training, all k-mers are encoded to obtain their latent representations z, which are clustered using a Gaussian mixture model (GMM). Cluster quality is assessed using the silhouette score. For visualization, UMAP projects the latent representations onto a two-dimensional space, where each k-mer is plotted and colored by its GMM cluster label. Each k-mer is associated with a theoretical modeling count and an empirical sequencing count; these counts are aggregated based on cluster labels. The correlation between these aggregates evaluates how high-dimensional structural features determine sequencing biases in RNA-seq.