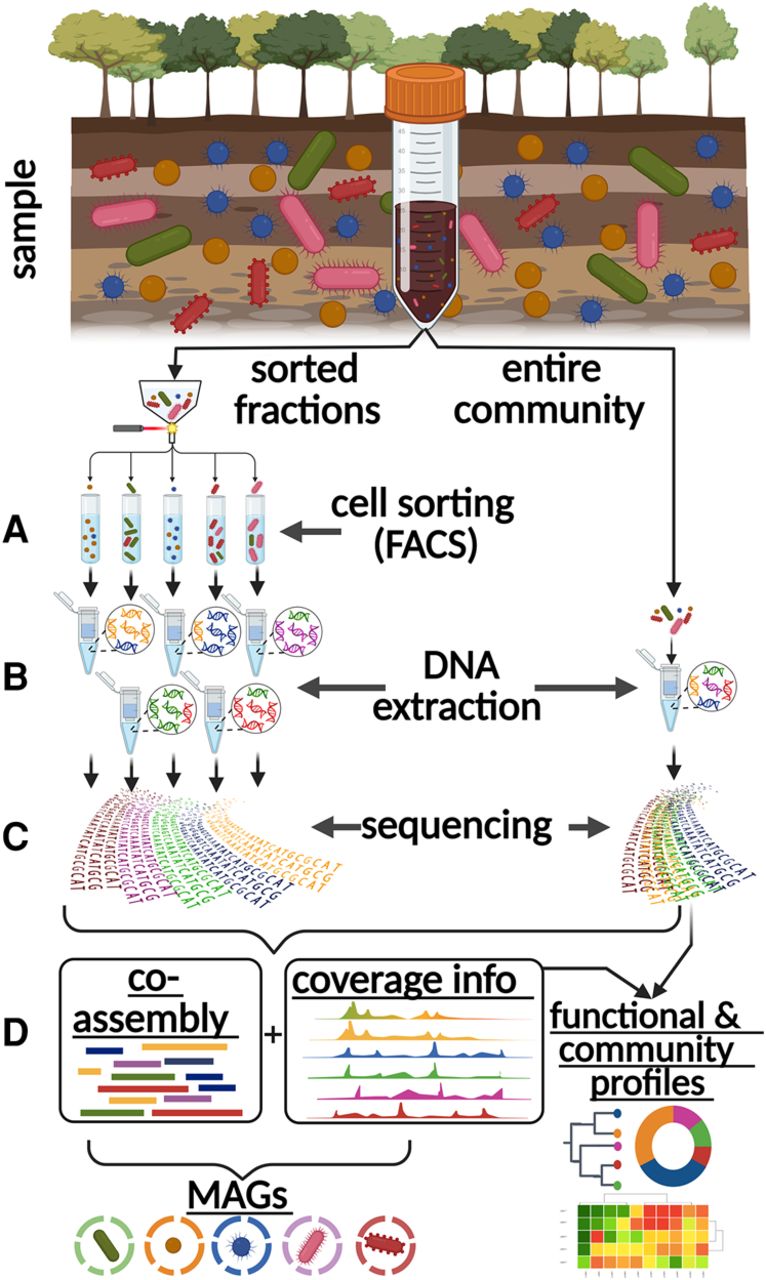
Midi-metagenomics workflow. (A) Part of the sample community is fractionated into distinct groups of several hundred thousand to millions of cells by cell sorting. (B) Different cell types are not separated with absolute stringency, but differentially enriched DNA is extracted separately from each fraction, as well as the original unsorted sample. (C) Extracted DNA is sequenced directly without whole-genome amplification (WGA). (D) Because the resulting read data sets represent different enrichments based on the same original community, they are optimal for coassembly as well as coabundance variation–based binning approaches. An unbiased representation of the source community is achieved by also including the original unsorted sample in the analyses. Created with BioRender (https://www.biorender.com).