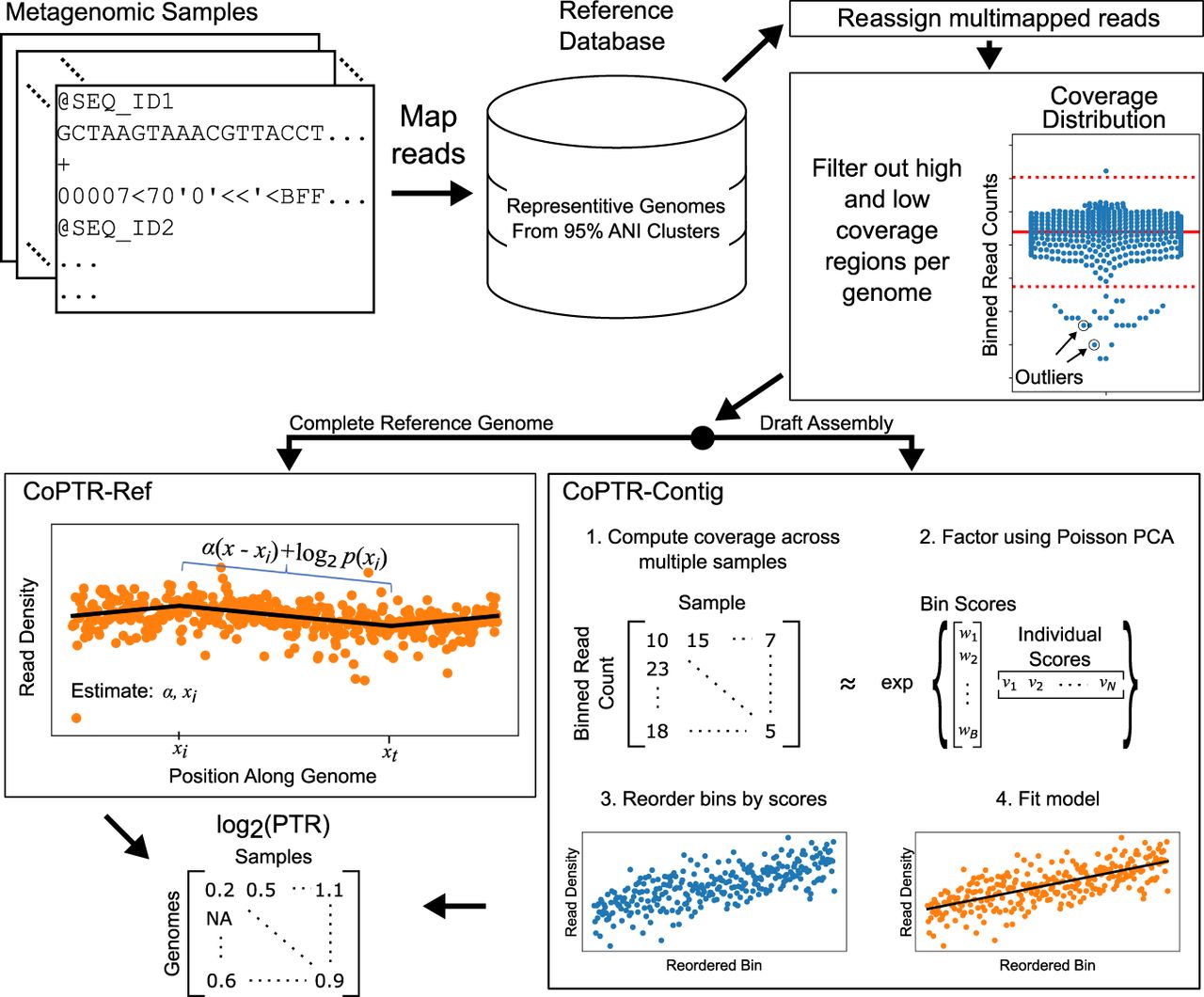
CoPTR workflow. Sequencing reads from multiple metagenomic samples are mapped to a reference database containing representative strains from complete reference genomes and high-quality assemblies (>90% completeness, <5% contamination). Multimapped reads are reassigned to a single genome using a probabilistic model. After read mapping, regions of each genome with ultra-high or ultra-low coverage are filtered using filters designed for complete reference genomes or draft assemblies. Then, PTRs are computed for each genome in each sample. For species with complete reference genomes, PTRs are estimated by maximizing the likelihood of a model describing the density of reads along the genome (CoPTR-Ref). For species with high-quality assemblies, reads are binned across the assembly, bins are reordered based on sequencing coverage across multiple samples using Poisson PCA, and the slope along this order is estimated by maximum likelihood (CoPTR-Contig). CoPTR outputs a table of the log2(PTR) per genome in each sample.