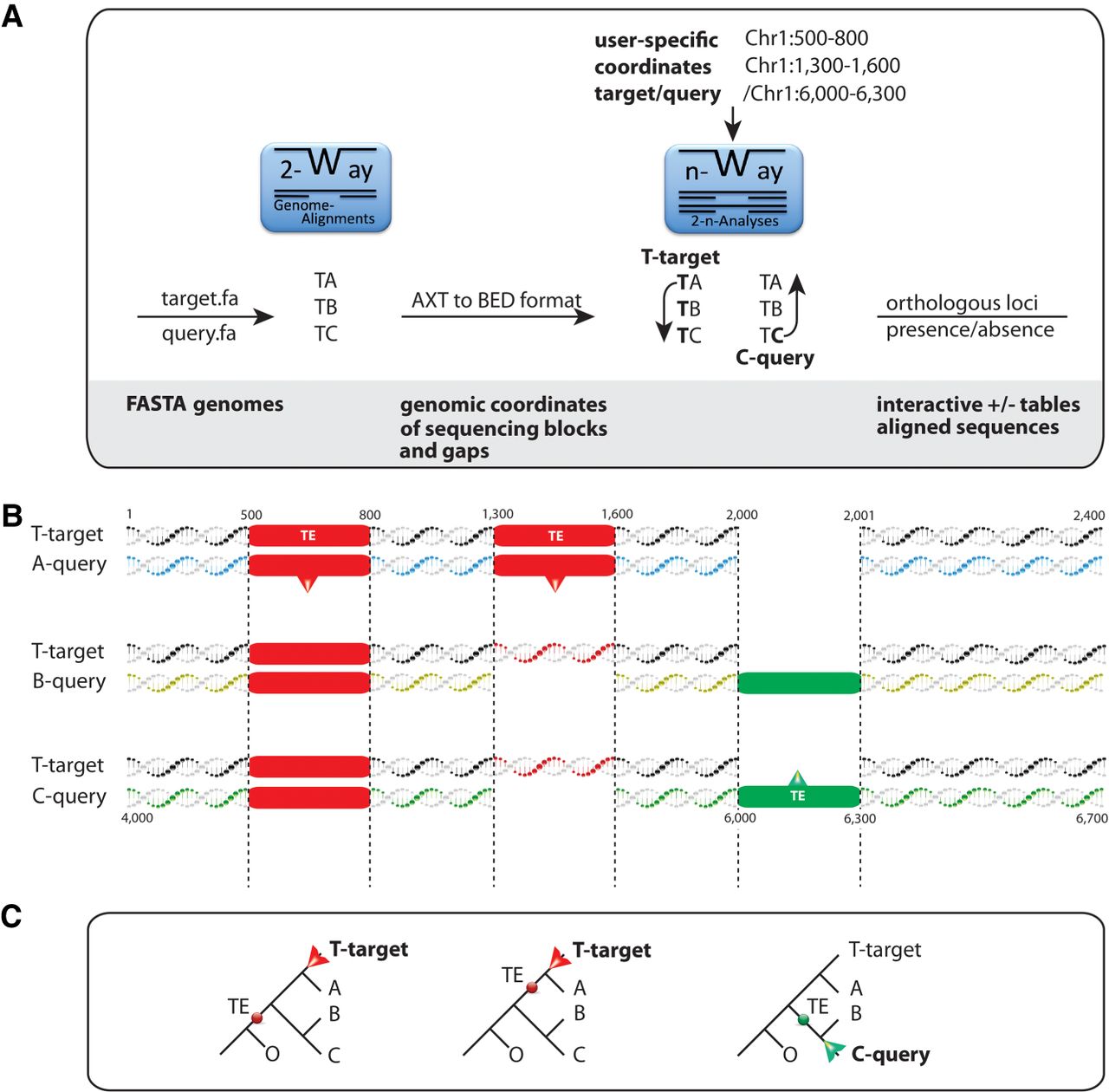
The 2-n-way suite. (A) The workflow from genome data to 2-way genome alignments and extraction of presence/absence patterns. 2-way: Input for pairwise alignments consists of assembled genomes at the chromosome- or scaffold-level (FASTA genomes). We distinguish between target genome assemblies (T; target.fa) as leading sequences in the generation of 2-way alignments and the query genomes (A, B, C; query.fa). The 2-way module generates a list of files, including AXT-formatted (https://genome.ucsc.edu/FAQ/FAQformat.html) coordinates and sequences that will be transformed into BED-formatted coordinates that can then be transferred to the n-way module. N-way: In n-way, all combinations of 2-way alignments are screened for specific coordinates of sequences present in the target (direct search) or the query (reverse search). (B) Visualization of direct and reverse searches at the sequence level from pairwise aligned genomic regions with labeled coordinates of transposable elements. The green-labeled TE is not recognizable via the coordinates of the target T but is identified by the coordinates of query C, and its presence is subsequently screened for in query B (reverse search). (C) Phylogenetic representation of the two search strategies. The direct search for diagnostic presence/absence markers (red dots) via target T (red arrow) recognizes the first TE shared by T, A, B, and C and the second TE present only in the target T and query A from the RepeatMasker coordinate table derived from the target. The reverse screening starts from the presence of a TE from query C (green dot; green arrow) based on the RepeatMasker coordinate table of query C that would not be recognizable in direct search.