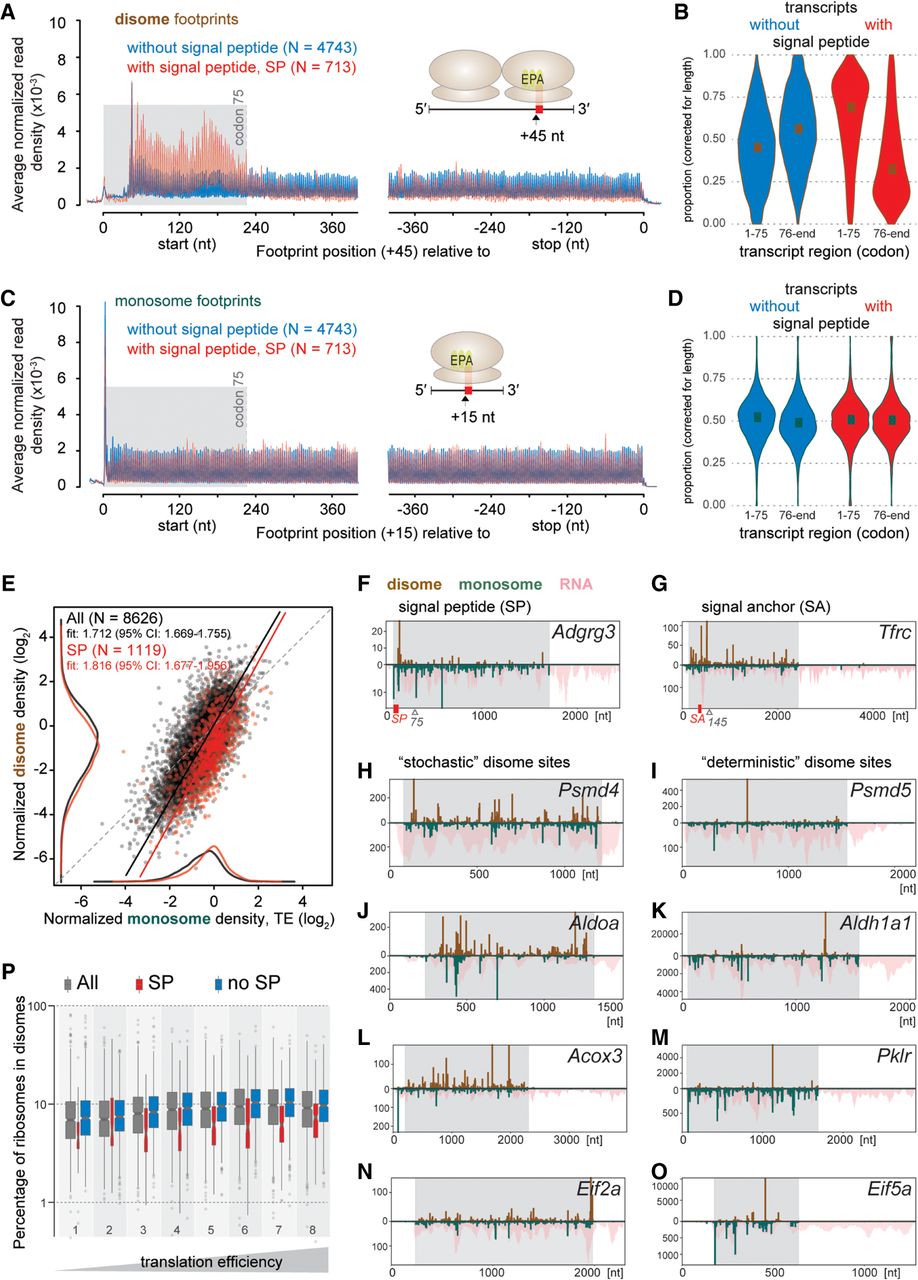
Disomes are associated locally with signal peptides and globally with high volumes of translation. (A,C) Density distribution of disome footprints identify signal peptide (SP)-related pausing events. Metatranscript analysis quantified the mean normalized footprint densities of disomes (A) and monosomes (C) within 400 nt from the start or −400 nt from the stop codons of transcripts encoding SPs (red, N = 713) or not (blue, N = 4743). (B,D) Violin-plots show the probability densities of length-normalized proportions of footprints within the first 75 codons and the rest of CDS from transcripts with (red, N = 713) or without (blue, N = 4743) SP for disomes (B) and monosomes (D). (E) Scatterplot of the relationship between per-gene normalized densities of disome and monosome footprints. All genes (N = 8626) were marked red or black depending on if they coded a SP (N = 1119) or not, respectively. Kernel density estimates are plotted on the margins (monosome on x-, disome on y-axis) for data sets of all genes (black) and SP coding genes (red) (without an axis of ordinates). Deming regression (errors-in-variables model) lines are shown for all genes (black) and the SP-coding subset (red). Regression slopes and their 95% confidence intervals (CI) are given in the top-left legends. Dashed gray line indicates the 1-to-1 slope. (F–O) Distribution of normalized counts of monosome and disome footprints along transcripts of representative genes confirms stochastic versus deterministic sites. The upward y-axis of the bar-plots shows the normalized read counts for disomes (brick red), while the downward y-axis was used for monosomes (teal) and total RNA (pink, pile-up). Transcript coordinates (nt) are shown on the x-axis; CDS regions are shaded in gray. If present, SP or signal anchor (SA) regions are indicated as red boxes along the x-axis. Plots show: Adgrg3, Tfrc, Psmd4, Psmd5, Aldoa, Aldh1a1, Acox3, Pklr, Eif2a, and Eif5a in F–O, respectively. (P) Box-plots illustrate the estimated proportion of ribosomes retained in disomes as a percentage of all translating ribosomes for different groups of genes. Box-and-whiskers were drawn for all genes detectable in the spike-in experiment (gray, N = 7375), subsets that code for SP (red, N = 892) or not (blue, N = 6483) and stratified into eight groups based on the octiles of the TE calculated from all genes, with right-closed interval boundaries (−5.41, −1.23, −0.77, −0.47, −0.23, −0.04, 0.17, 0.47, 3.17), depicted as increasing TE below the graph. Width of each box is proportional to the number of data points it represents.