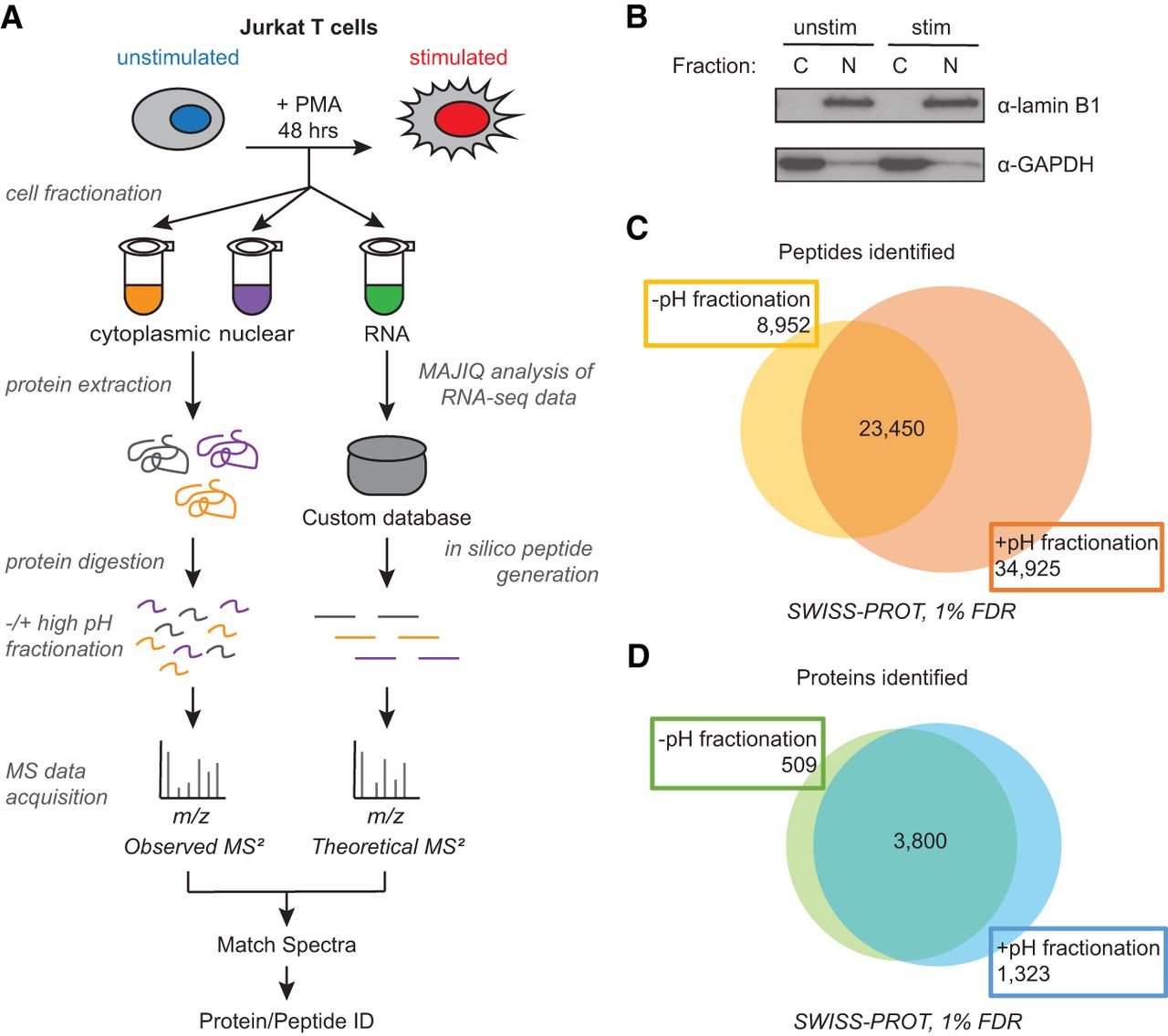
Increased protein and peptide identification achieved by high pH peptide fractionation. (A) Sample processing workflow used for integration of mass spectrometry (MS)–based proteomics and RNA-seq data. Jurkat T cells (JSL1) were stimulated for 48 h with phorbol 12-myristate 13-acetate (PMA) before protein or RNA extraction. Cells were harvested and fractionated into cytoplasmic and nuclear subfractions for MS analysis. Additionally, a second fractionation step was introduced after protein digestion to decrease sample complexity (high pH peptide fractionation). MS data were acquired via data-dependent and data-independent acquisition (DDA and DIA). Right path in workflow is described in Figure 1. (B) Validation of subcellular fractionation by western blot using antibodies for GAPDH and lamin B1. (C) Number of peptides identified −/+ high pH peptide fractionation at 1% false-discovery rate (FDR). (D) Number of proteins identified −/+ high pH peptide fractionation.