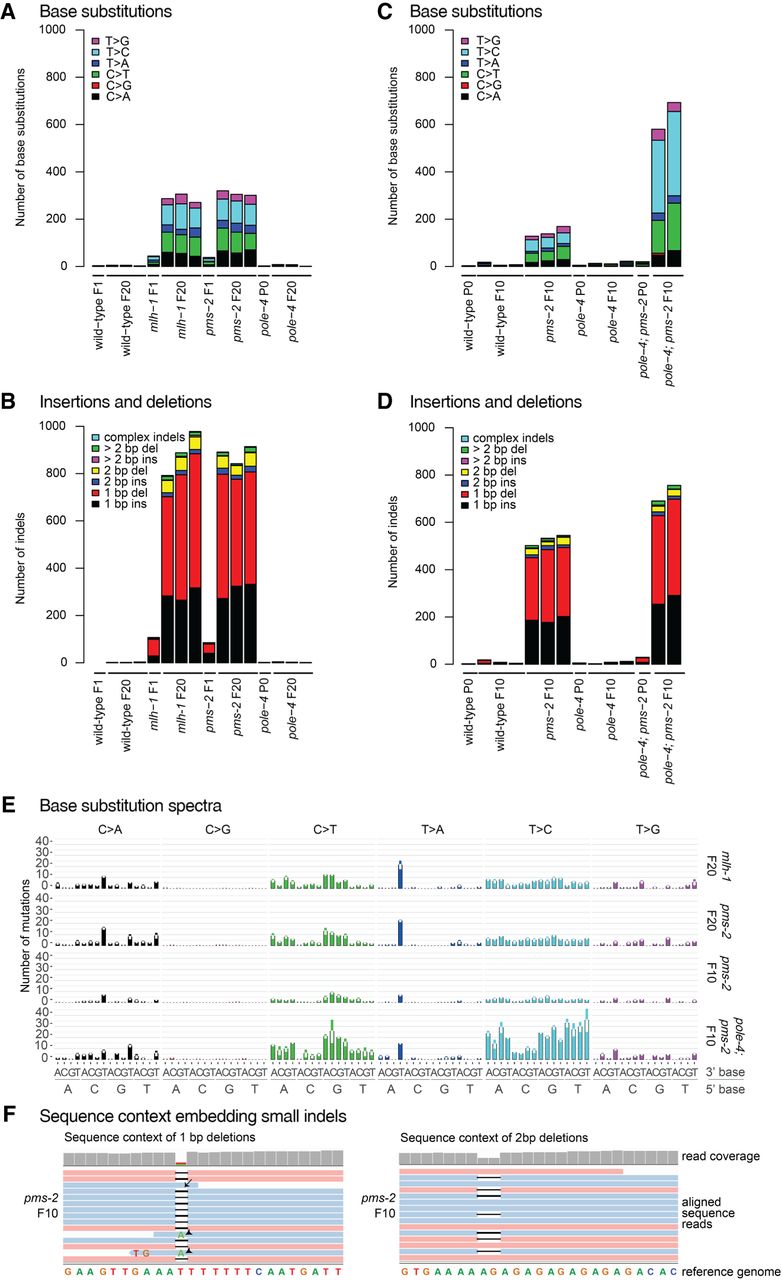
Mutations in C. elegans wild-type and MMR mutants grown for 10 or 20 generations. Identical base substitutions as well as indels occurring in the same genomic location among samples of the entire data set (duplicates) were excluded from the analysis, thus only reporting mutations unique to each individual sample. (A) Number and types of base substitutions identified in the parental (P0) or one first generation (F1) line and three independently propagated F20 lines of wild-type, mlh-1, pms-2, and pole-4 single mutants. (B) Number and types of insertions and deletions (indels) identified in initial (P0 or F1) and three independently propagated F20 lines of wild-type, mlh-1, pms-2, and pole-4 single mutants. (C) Number and types of base substitutions observed in the parental (P0) line and 2–3 independently propagated F10 lines of wild-type, pms-2 and pole-4 single, and pole-4; pms-2 double mutants. (D) Number and type of indels observed in the parental (P0) and 2–3 independently propagated F10 lines of wild-type, pms-2 and pole-4 single, and pole-4; pms-2 double mutants. (E) Average number of base substitutions identified across all individual lines per genotype in their 5′ and 3′ base sequence context in mlh-1 and pms-2 single and in pms-2 single and pole-4; pms-2 double mutants. Error bars represent the standard error of the mean. (F) Examples of indel sequence contexts. Sequence reads aligned to the reference genome WBcel235.74 visualized in Integrative Genomics Viewer (Robinson et al. 2011). A 1-bp (left) and a 2-bp deletion (right) are shown. A subset of sequence reads, which end close to an indel, erroneously aligned across the indel resulting either in wild-type bases (arrow) or base changes (arrowheads). Such wrongly called base substitutions were removed during filtering (Methods) using the deepSNV package (Gerstung et al. 2012, 2014).