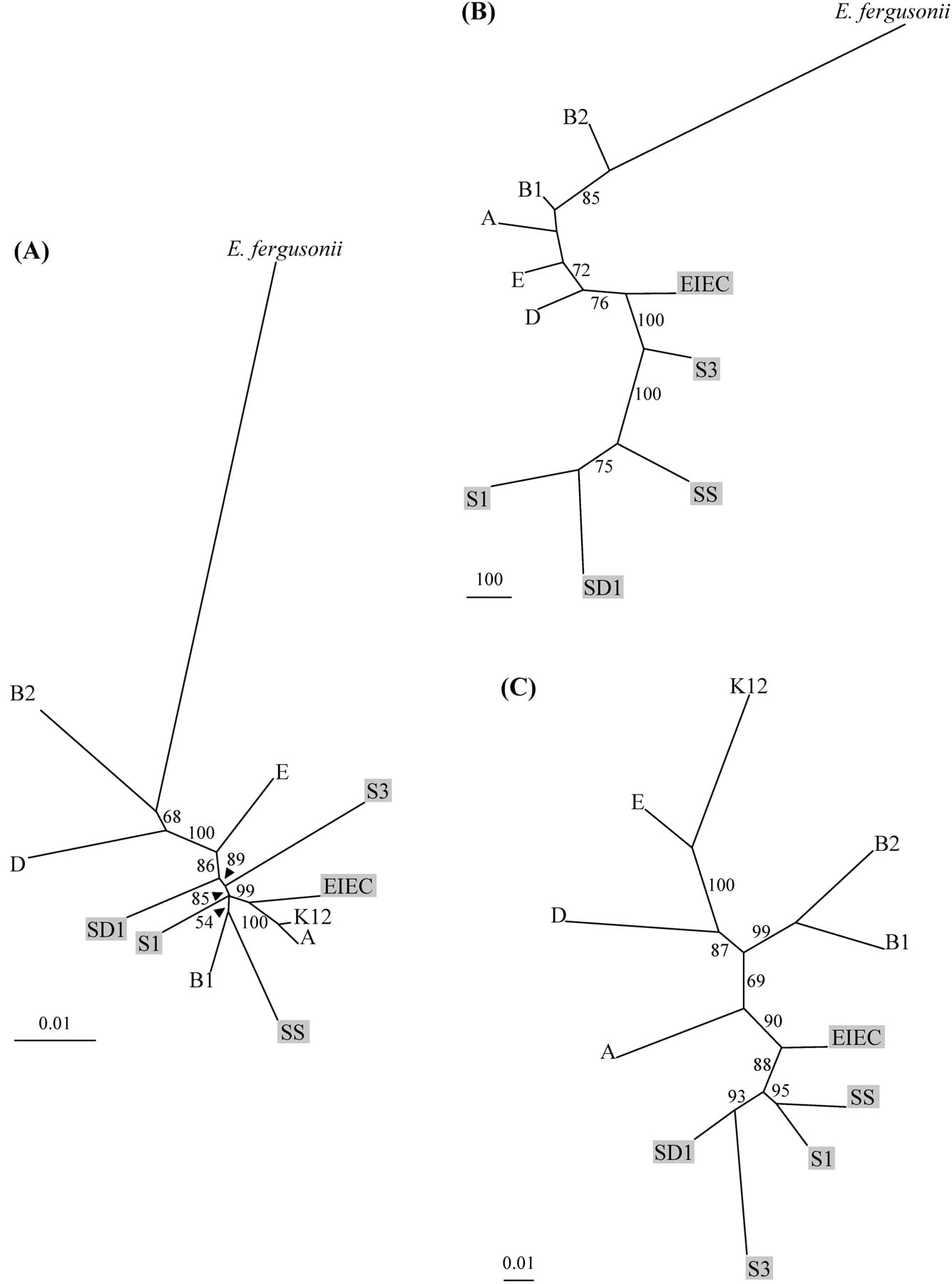
(A) Phylogenetic tree of the 12 strains, E. fergusonii as an outgroup, and reconstructed from the DNA sequences of 11 essential genes, using Neighbor Joining procedure. The model used to estimate the pairwise distance matrix is TN93, the parameters being estimated by maximum likelihood (PHYML [Guindon and Gascuel 2003]) as follows: transition/transversion for purines = 3.28, transition/transversion for pyrimidines = 6.08, γ shape = 0.112, 10 categories, percentage of invariant = 0. Identical topologies were obtained using maximum likelihood or parsimony procedures (data not shown). (B) Phylogenetic tree of the 11 strains (K12-MG1655 being excluded), E. fergusonii as an outgroup, and obtained by parsimony procedure using PAUP4.0b (Swofford 2002) from the K12-MG1655 4290 binary-coded ORFs (“0” for undetectable sequence, “1” for sequence present at least in one copy). The tree is obtained by branch and bound algorithm, 1264 characters being parsimony informative. The length of the tree is 3584 steps, consistency index = 0.56, retention index = 0.52. (C) Unrooted tree of the transcript abundances, expressed as adjusted RNA values (see Methods), of the 2880 core genome genes for the 11 strains (E. fergusonii excluded). The tree is obtained following the Neighbor Joining algorithm applied to the euclidian distances between strains. All of the bootstrap values are obtained from 1000 replicates and are indicated only when ≥50%. Abbreviations for the strain designation are as follows: B2 (ECOR56), B1 (ECOR26), D (ECOR50), E (EDL933), A (ECOR1), K12 (K12-MG1655), EIEC (EIEC85b), SD1 (S. dysenteriae serotype 1, SD0177), SS (S. sonnei, SS92a), S1 (S. boydii serotype 10, SB1080), S3 (S. flexneri serotype 5, M90T). Shigella and EIEC strains are indicated in gray background.