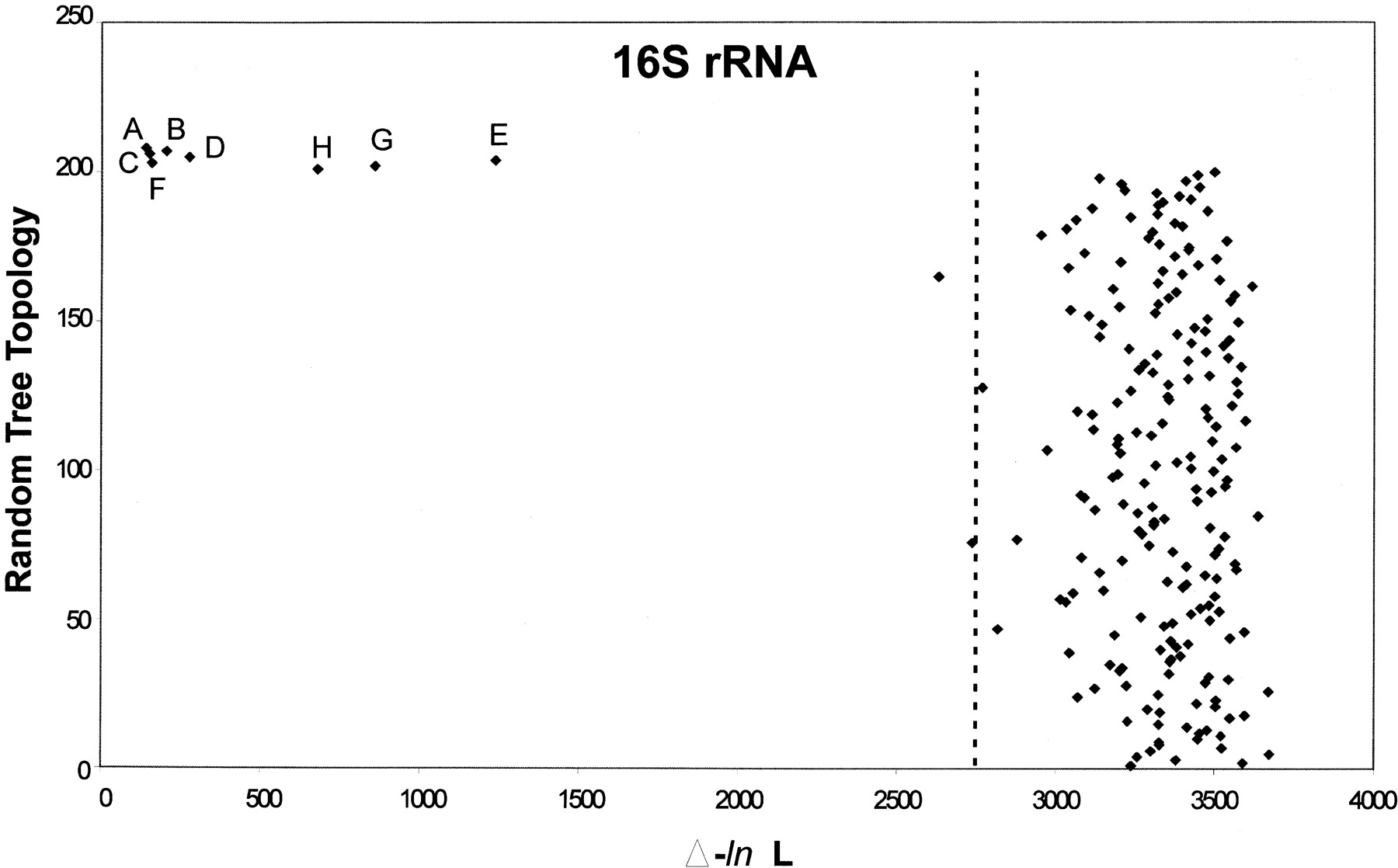
Likelihood analysis of phylogenetic congruence in prokaryotes. The phylogeny based on 16S rRNA is compared with phylogenies based on RpoA, GroE, RecA, whole-genome dinucleotide usage deviation, whole-genome tetranucleotide usage deviation (TUD), coding DNA TUD, or noncoding DNA TUD. The letters represent the locations of the distances in log likelihood (Δ-ln L) between the 16S rRNA phylogeny and: RpoA (A), GroE (B), RecA (C), whole-genome TUD based on zero-order Markov criteria (D), whole-genome TUD based on Markov chain analysis (E), coding DNA TUD based on zero-order Markov criteria (F), noncoding DNA TUD based on zero-order Markov criteria (G), and whole-genome dinucleotides based on zero-order Markov criteria (H). The 99th percentile of the likelihood differences between the 16S rRNA tree and the topologies from 200 random trees is indicated by the dotted line.