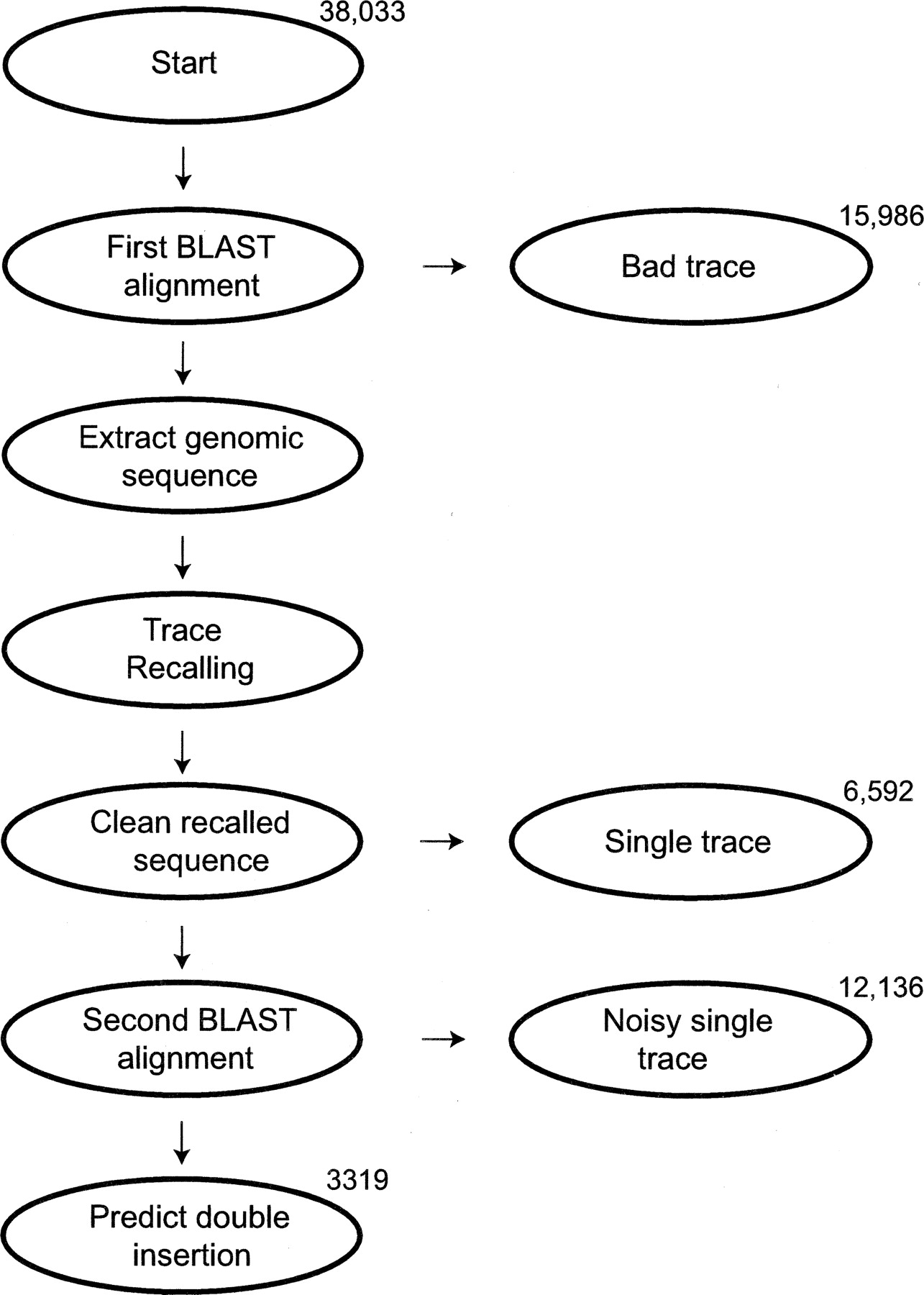
Diagram of pipeline used to analyze the Arabidopsis random insertional mutagenesis experiment. Numbers of traces present at key stages of the pipeline are noted. The trace is first called without ambiguity codes using PHRED with default parameters. This sequence is aligned to the whole Arabidopsis genome using BLAST. If there is no significant alignment, the trace is discarded. Otherwise, the aligned genomic sequence plus 1000 bases of flanking sequence on either side of the alignment are extracted. This is assumed to be the locus of one insertion event. Trace Recalling is applied to this extracted genomic segment and the trace. If the trace is a double trace resulting from two insertion events, the recalled sequence is a chimera of the T-DNA sequence, the genomic sequence flanking the second insertion, and the single-trace portion of the sequence flanking the first insertion. Therefore, in the next step we remove single-trace segments of the sequence by removing any subsequences of the recalled sequence that align well to the originally called sequence. This is called cleaning the recalled sequence. If this step removes all of the recalled sequence, the trace is classified as a single-insertion event and removed from the pipeline. Otherwise, the remaining recalled sequence is aligned to the genome with BLAST. If this alignment is not significant, we assume the recalled sequence represents noise and the trace is classified as a noisy single trace. However, if the alignment is significant, we call the trace as a double trace representing two insertion events and predict the locus of the second insertion as the location of the second BLAST hit.