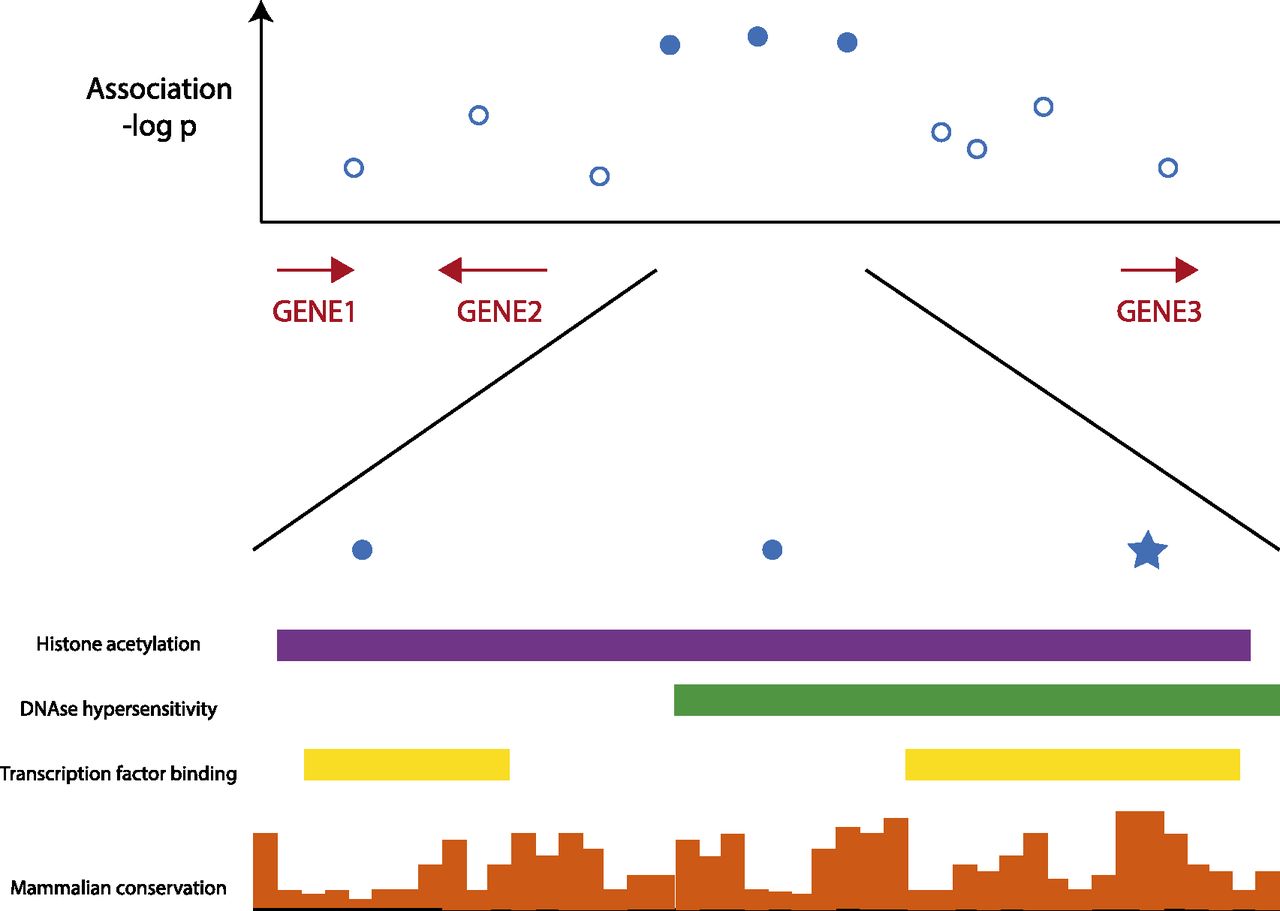
Hypothetical data showing how conservation can be used to identify the most likely functional variants in a disease locus. GWAS or sequencing data can be combined with constraint information and other genome annotations in the appropriate cell types to assess SNPs and other variants present on human disease-associated haplotypes. From the multitude of associated variants, one or a few candidate mutations often stand out based on the overlap of a candidate SNP (best variant indicated by star in lower panel) with constraint and other genome annotation. In a best-case scenario, careful analysis of sequence motifs overlapping the candidate variant will also identify a transcription factor binding site, splice site, or RNA structure that is altered by the candidate variant.