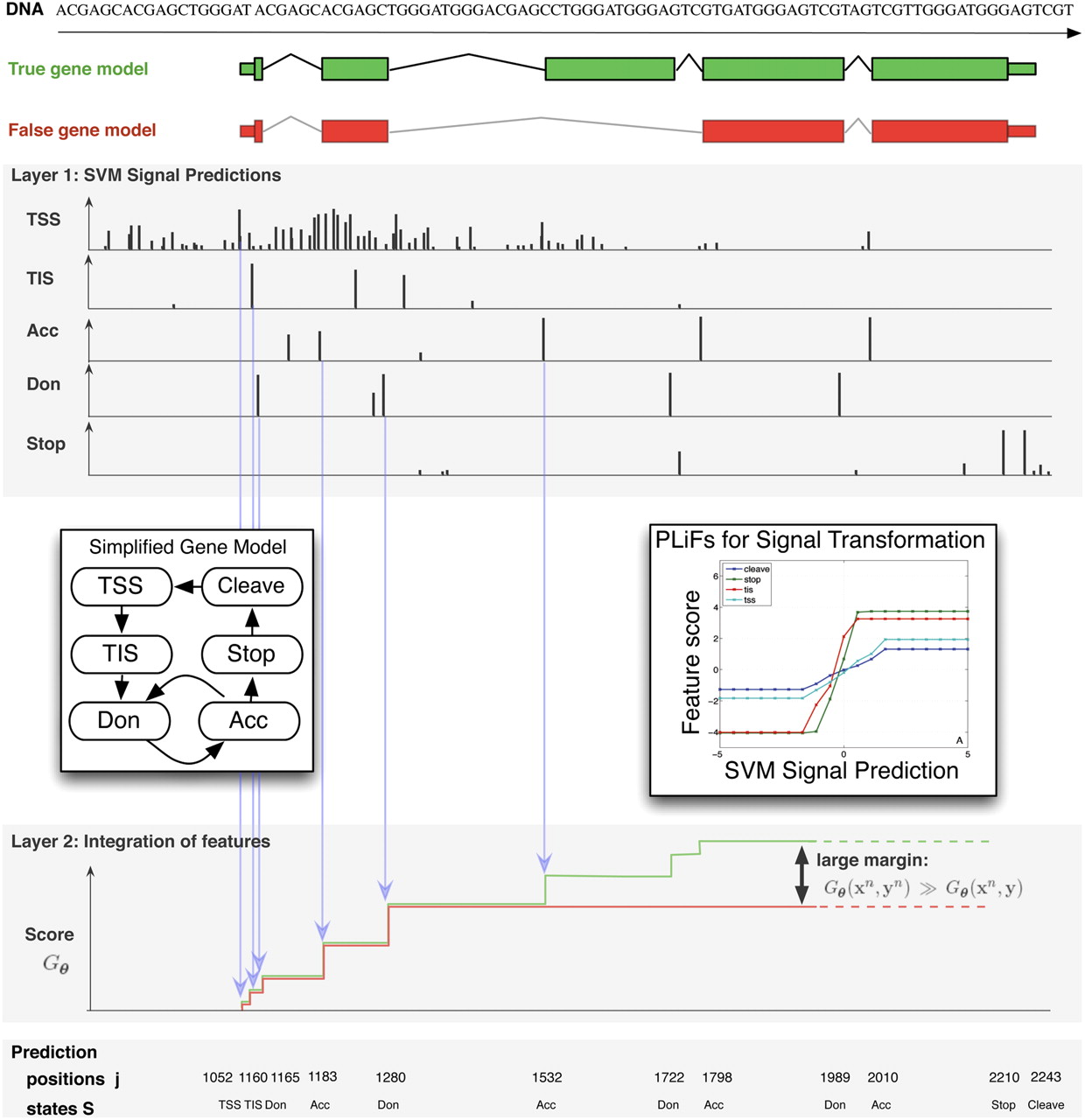
In layer 1, mGene scans the genomic sequence using SVM-based detectors trained to recognize transcription start sites (TSS), translation initiation sites (TIS), acceptor (Ace), and donor (Don) splice sites, the translation termination site (Stop), and other signals (data not shown). The detectors assign a score to each candidate site. In combination with additional information, including outputs of SVMs recognizing exon/intron content, and scores for exon/intron lengths (data not shown), these signal scores contribute to the cumulative score of a putative gene structure. The bottom graph (layer 2) illustrates the accumulation of scores for two gene structures shown at the top, where the score at the end of the sequence is the final score of the gene structure. The contributions from the individual detector outputs, from segment lengths, as well as from properties of the segments to the score are adjusted during training using piecewise linear functions (PLiFs; see inset to the right). They are optimized such that the margin between the true gene structure (shown in green) and all other (false) isoforms (one of them is shown in red) is maximized. Prediction of genes on new sequences works by selecting a valid gene structure, as defined by the gene model (cf. inset to the left), with the maximum cumulative score using dynamic programming (see e.g., Kulp et al. 1996).