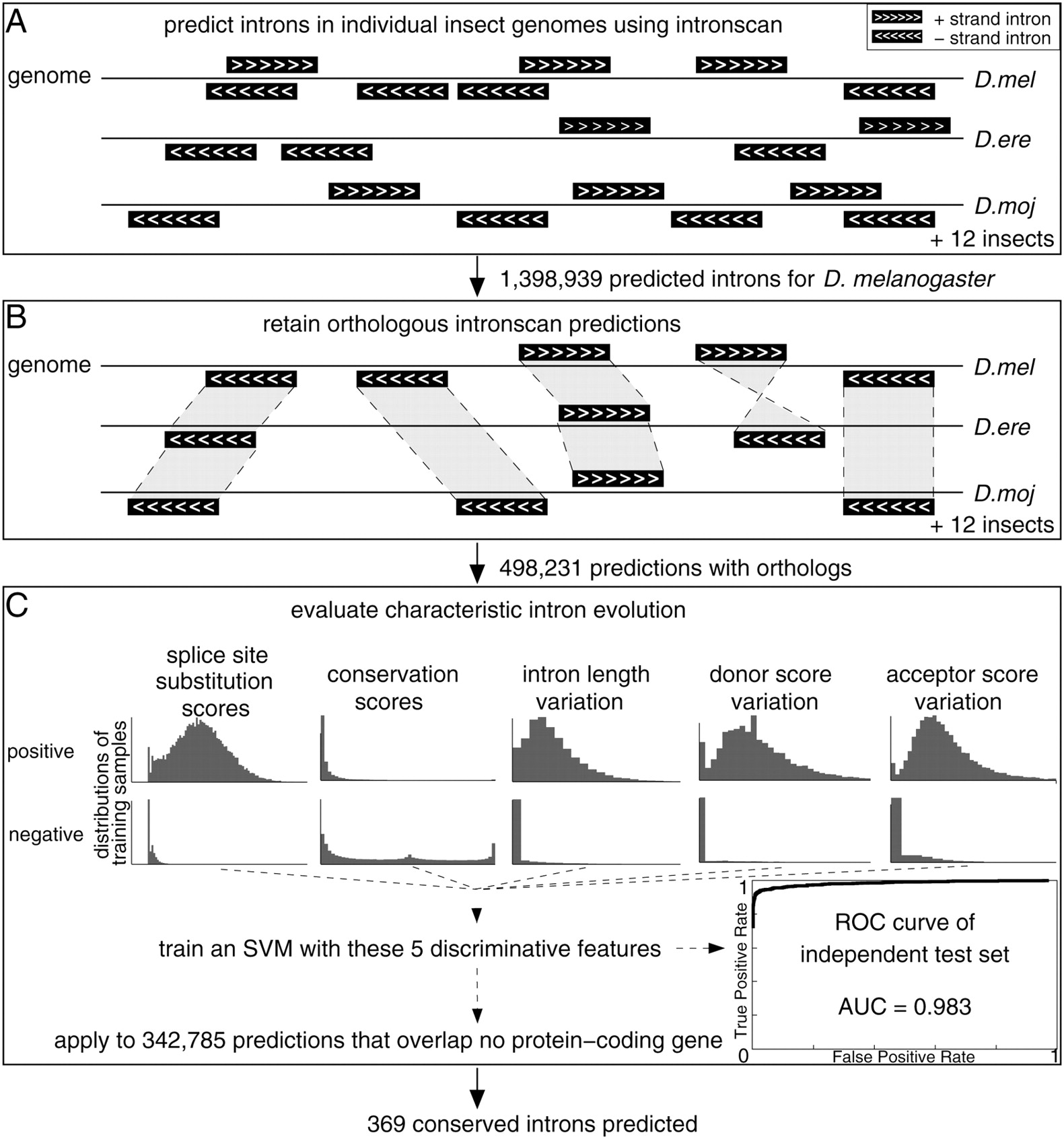
Overview of the computational intron prediction procedure. (A) Introns are predicted using intronscan on both strands of the D. melanogaster genome, yielding a total of ∼1.4 million predictions. Independent intronscan predictions in the other insect genomes were made. (B) Only those D. melanogaster intron predictions are retained that have an orthologous prediction in at least one additional genome. (C) A support vector machine (SVM) classifier based on five features is used to distinguish positive (real introns) and negative training samples (false predictions). These features measure characteristic splice site substitutions, sequence conservation in the middle part of introns, and variation of the intron length, donor, and acceptor score between species. As indicated by the distributions, these features are highly discriminative for positive and negative samples. By using this classifier, we predict 369 conserved introns.