Abstract
Genetic diversity within the human immunoglobulin heavy-chain (IGH) locus influences the expressed antibody repertoire and susceptibility to infectious and autoimmune diseases. However, repetitive sequences and complex structural variation pose significant challenges for large-scale characterization. Here, we introduce a method that combines Oxford Nanopore Technologies ultra-long sequencing and adaptive sampling with a bioinformatic pipeline to produce haplotype-resolved, annotated IGH assemblies. Notably, our strategy overcomes prior limitations in phasing resolution, enabling single-contig haplotype assemblies that span the entire IGH locus. We apply this method to four individuals and validate the accuracy of the IGH assemblies using Pacific Biosciences HiFi reads, demonstrating near-complete sequence congruence, with only some residual indel errors. Moreover, when applied to the reference material HG002, our pipeline reveals no base differences and a limited number of indels compared with the telomere-to-telomere genome benchmark across the IGH region. Importantly, in the four individuals, our approach uncovers 28 novel alleles and previously uncharacterized large structural variants, including a 120 kb duplication spanning IGHE to IGHA1 within the IGH constant region (IGHC) and, within the IGHV region, an expanded seven-copy IGHV3-23 gene haplotype. These findings underscore the power of our method to resolve the full complexity of the IGH locus and uncover previously unrecognized variants that may affect immune function and disease susceptibility. Thus, our method provides a strong basis for future immunological research and translational applications.
Immunoglobulins (Igs) are highly diverse effector molecules integral to adaptive immunity. They play crucial roles in defending against infectious agents but can also contribute to the development of autoimmune diseases. Initially expressed as surface B cell receptors (BCR), Igs recognize and bind antigens, thereby activating the B cells. This activation drives the differentiation into memory B cells and effector B cells, including plasmablasts and plasma cells, which secrete soluble Igs, or antibodies (Cyster and Allen 2019).
The basic structure of an Ig molecule consists of two identical heavy chains and two identical light chains, forming two heavy-light chain pairs. The Ig heavy chains (IGHs) are encoded within the IGH locus, an ∼1.5 Mb region located at the telomeric end of the long arm of Chromosome 14. The locus contains more than 100 homologous gene segments divided into four classes: constant (C), variable (V), diversity (D), and joining (J) genes (Lefranc and Lefranc 2001). The C genes encode the constant region of the antibody, determining the effector functions, whereas the V, D, and J genes encode the variable domain responsible for antigen binding. The IGH locus exhibits high levels of structural variation (SV) and allelic diversity, contributing to significant variability in the germline repertoire within a population and between the haplotypes in the same individual (Rodriguez et al. 2023). During B cell development, allelic exclusion ensures that only one IGH haplotype is expressed in each B cell. This occurs after successful V(D)J recombination on one chromosome, preventing further recombination on the other chromosome (Roldán et al. 2005). A highly diverse expressed repertoire is generated through various combinations of V, D, and J gene segments, as well as additions and deletions at their junctions (Feeney 1992).
Increasing evidence suggests that variation in the germline IGH locus significantly affects the expressed Ig repertoire (Mikocziova et al. 2021). Studies in monozygotic twins indicate that key features in the expressed antibody repertoire are heritable (Wang et al. 2015; Rubelt et al. 2016), and recent research shows that IGH germline polymorphisms in both coding and noncoding regions influence the V, D, and J gene usage frequencies on a population scale (Rodriguez et al. 2023). Furthermore, conserved germline-encoded residues that influence antibody specificity and V, D, and J gene usage have been demonstrated in the immune response against pathogens such as influenza A virus (Avnir et al. 2016), HIV (Scharf et al. 2013; Yacoob et al. 2016; deCamp et al. 2024), Staphylococcus aureus (Yeung et al. 2016), and Plasmodium falciparum (Tan et al. 2018). Recent studies have also uncovered considerable unexplored germline variation within the IGHC genes (Calonga-Solís et al. 2019; Bashirova et al. 2021; Ford et al. 2023). Polymorphisms in the IGHC region can influence Fc receptor binding and, thereby, antibody effector functions (Ternant et al. 2016; De Taeye et al. 2020). In membrane-bound IgG, a newly discovered variant in the IGHC region encoding the intracellular IgG1 tail was shown to modulate B cell activation and differentiation and was also a risk variant for systemic lupus erythematosus (Chen et al. 2018). In multiple sclerosis (MS), B cells expressing specific IGHG1 gene variants are selected for in the central nervous system (Lossius et al. 2017) and correlate with increased intrathecal antibody synthesis (Buck et al. 2013).
Our present knowledge of genetic variation within the IGH locus remains incomplete. The high degree of polymorphism, repetitive sequences, copy number variations (CNVs), and SVs have posed significant challenges in characterizing the IGH locus on a large scale (Watson et al. 2015). As a result, IG genes have largely been excluded from genome-wide association studies in autoimmune and infectious diseases (Watson and Breden 2012). Encouragingly, recent technological advancements have enabled the generation of longer sequencing reads with increased accuracy. A framework using capture probes and highly accurate Pacific Biosciences (PacBio) HiFi long-read sequencing allows for high-throughput characterization of the locus, and efforts are underway to gather germline diversity across human populations (Rodriguez et al. 2020). However, although this strategy yields highly accurate reads, it does not contiguously resolve the entirety of the locus and may miss larger SVs. Oxford Nanopore Technologies (ONT) whole-genome sequencing offers increased contiguity of assemblies and improved phasing over longer stretches owing to read lengths (Beaulaurier et al. 2025) but has been hampered by lower raw read accuracy, necessitating deeper sequencing (Ni et al. 2023).
To address these challenges, we aimed to establish a protocol for generating phased and complete haplotype-resolved IGH assemblies using only ONT ultra-long sequencing. In this study, we evaluate the effectiveness of the proposed method and the quality of the resulting assemblies and compare them to those of an orthologous PacBio method. Additionally, we highlight the utility of the method by describing novel genes and large structural variants, thereby advancing our understanding of the IGH locus and basic human immunogenetics.
Results
Generating single-contig haplotype-resolved IGH assemblies
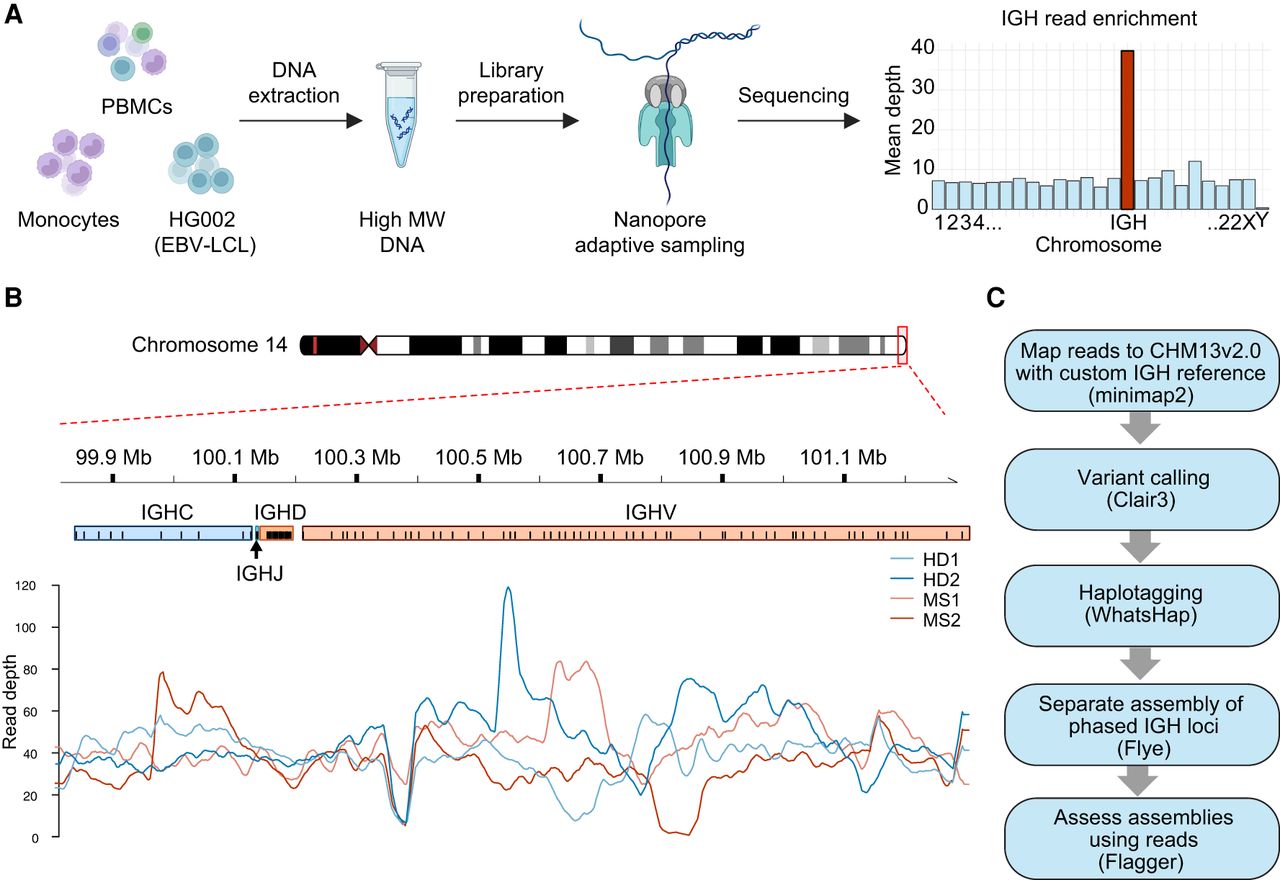
We have established a protocol for comprehensive characterization of the IGH locus using ONT ultra-long sequencing with adaptive sampling (Loose et al. 2016). This method was validated on samples from four donors—two healthy individuals (HD1 and HD2) and two MS patients (MS1 and MS2)—and the Epstein–Barr virus transformed lymphoblastoid cell line (EBV-LCL) HG002. The donors represent diverse ethnic backgrounds: The healthy donors are of African American and Asian ancestry (HD1 and HD2, respectively), and the MS patients are of European and South Asian ancestry (MS1 and MS2, respectively). We established a sequencing protocol with ONT adaptive sampling, in which reads from our region of interest (ROI) were enriched without the need for target amplification during library preparation (Fig. 1A). This approach yielded a mean read depth of approximately 30× over the IGH locus (Fig. 1B), representing a five- to sevenfold increase compared with the mean depth across the genome (Table 1). Furthermore, we obtained ultra-long reads with maximum read lengths exceeding several hundred kilobases, high read N50 values ranging from 65 to 95 kb, and read median Phred scores between 20.3 and 21.6 (Table 1; Supplemental Fig. S1).
Overview of the experimental setup and bioinformatic pipeline. (A) Schematics of DNA sample preparation and sequencing. High-molecular-weight gDNA was extracted from peripheral blood mononuclear cells (PBMCs), isolated monocytes, or the HG002 Epstein–Barr virus lymphoblastoid cell line (EBV-LCL). The DNA was used as input for the preparation of ultra long-read ONT sequencing libraries. Sequencing was performed using an adaptive sampling strategy to enrich for reads mapping to the IGH locus. (B) Read depth from the four donors on a custom IGH reference. (C) Outline of the bioinformatic pipeline used to generate one-contig haplotype-resolved IGH assemblies.
Descriptive statistics on ONT data input to make IGH assemblies
| Individual | Longest IGH read (bp) | IGH N50 (bp) | Mean genome depth | Mean IGH depth | Fold enrichment | Median Phred score | Proportion bp in >100 kb IGH reads |
|---|---|---|---|---|---|---|---|
| HD1 | 420,227 | 92,574 | 5.3 | 33.6 | 6.3 | 21.7 | 45.9% |
| HD2 | 270,970 | 81,463 | 7.0 | 39.8 | 5.7 | 20.7 | 39.8% |
| MS1 | 358,847 | 66,201 | 6.7 | 40.0 | 6.0 | 20.3 | 32.4% |
| MS2 | 372,947 | 74,433 | 3.9 | 28 | 7.2 | 20.7 | 34.6% |
| HG002 | 287,836 | 79,220 | 6.2 | 24.9 | 4.0 | 19.8 | 37.9% |
Using these data, we developed a bioinformatic pipeline employing existing tools (Fig. 1C), which enabled the generation of high-quality, contiguous haplotype-resolved assemblies. The pipeline utilizes a custom IGH reference that incorporates several common SVs to enable reference-based phasing of reads. Next, the phased reads are separately assembled de novo. We assessed the accuracy of the initial haplotype-resolved assembly drafts, which allowed us to correct phase shifts or revise potential misassemblies (see below). The final corrected IGH loci in all four donors and in HG002 were assembled in single contigs without any apparent phasing ambiguities.
Assessing the accuracy of IGH assemblies
Large duplications pose a challenge for de novo assembly, especially in cases with high sequence similarity between duplicated segments. These SVs are particularly common at this locus, making it crucial to evaluate the assembly and correct any inaccuracies. To ensure the quality of our IGH assemblies, we employed several strategies for rigorous quality control. First, we used the Flagger pipeline to assess assembly reliability (Liao et al. 2023). In this approach, reads are mapped back to the corresponding IGH draft assemblies, and coverage distribution is analyzed to identify potential misassemblies. Three of the draft diploid assemblies showed indications of collapsed or erroneous sequences (Table 2). We verified true errors by checking for distinct patterns of single-nucleotide variants (SNVs) in the read alignments, or by identifying clear boundaries with soft-clipped reads surrounding the region. If assembly errors were confirmed, we manually corrected them, before reevaluating the new assembly. The size and type of errors detected in the different draft assemblies varied between the donors, which reflects the individual donors’ unique SVs. In the final IGH assemblies, only a few bases remained flagged as misassembled, none of which could be resolved upon manual inspection. These unconfirmed flagged errors represent either assembly errors too large for our reads, or artifacts owing to uneven read coverage or mapping.
Flagger evaluation of draft and finished haplotype-resolved IGH assemblies for the four donors
| Donor | HD1 | HD2 | MS1 | MS2 | ||||
|---|---|---|---|---|---|---|---|---|
| Draft | Finished | Draft | Finished | Draft | Finished | Draft | Finished | |
| Haploid | 2,835,084 | 3,067,029 | 2,633,025 | 2,800,672 | 2,771,599 | 2,945,333 | 1,400,097 | 1,400,097 |
| Collapsed | 51,043 | 0 | 125,554 | 35,052 | 84,321 | 0 | 246 | 246 |
| Duplicated | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Error | 0 | 0 | 23,793 | 0 | 40,884 | 0 | 0 | 0 |
[i] Table shows the number of bases of the dual assembly assigned to each of the four categories.
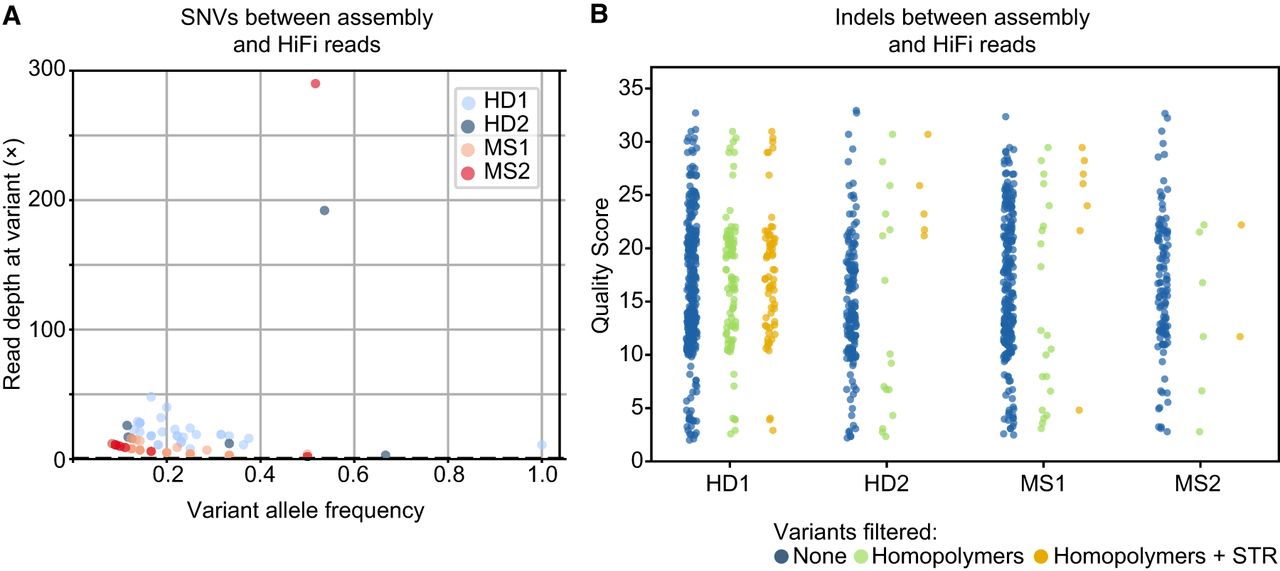
Additionally, we sequenced samples from our four donors using PacBio HiFi technology and used these high-accuracy reads to assess the sequence precision of our ONT assemblies. The HiFi reads were mapped to the final IGH assemblies generated with ONT data, and variants were called using Clair3. No definitive SNVs with high read coverage and allele frequency >0.8 were found (Fig. 2A). However, three SNVs of note were still detected. One SNV with allele frequency of one was called in HD1, but in an area with relatively low HiFi read depth (11×). Two high-coverage SNVs were called with frequency ∼0.5 in HD2 and MS2, indicating either true SNVs or misalignments. Indel errors between assemblies and HiFi reads were more numerous than SNVs. In total, 734 indels of quality greater than one were called (Fig. 2B) in the collective assemblies >10 Mb, giving a rate of 7.2 indels per 100 kb. The majority of these (n = 612) were found within ±1 bp of a homopolymer ≥5 bp.
ONT assemblies are concordant with HiFi reads Variant caller Clair3 estimated single-nucleotide variants (SNVs; A) and insertions/deletions (indels; B) between the donors’ HiFi reads and their haplotype-resolved IGH assembly from ONT data. (A) Scatter plot of HiFi read depth and variant allele frequency for SNVs from all donors. (B) Dot plot of quality score of indels called, stratified by different filtration. No filter (blue), indels within ±1 bp of homopolymers ≥5 bp (green), and indels within homopolymers or short tandem repeats of dinucleotide of seven or more repeats (orange).
Benchmarking with HG002
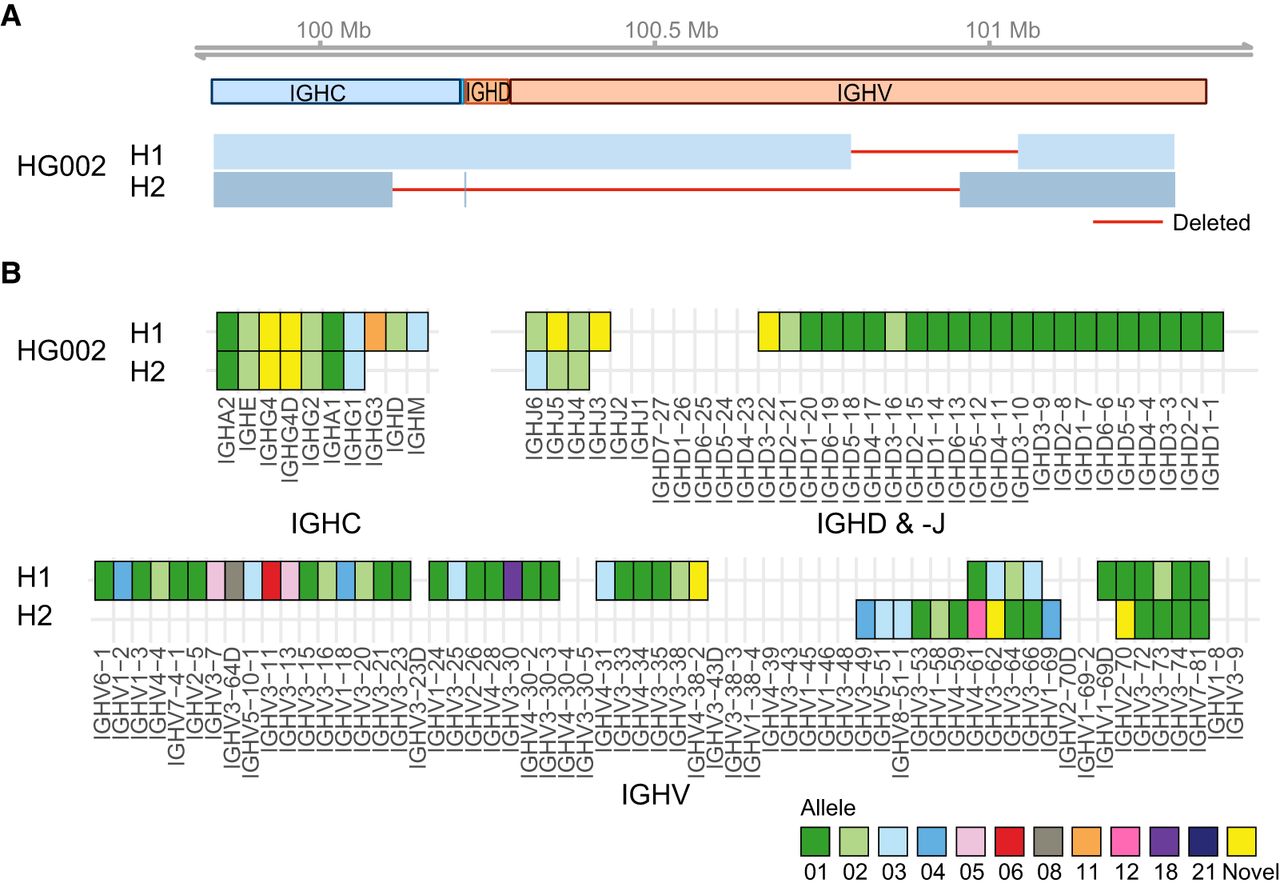
To further evaluate the performance of our long-read ONT method, we sequenced and assembled the IGH locus for the Genome in a Bottle (GIAB) HG002 reference material. HG002, also known as NA24385, is a thoroughly sequenced reference genome with extensive publicly available high-quality data sets with trio validation. Among these is the recently published telomere-to-telomere (T2T-GIAB Q100) complete diploid HG002 genome (Rautiainen et al. 2023). Using our method, we sequenced and assembled the HG002 IGH locus, resulting in one contig per haplotype. These assemblies were concordant with the Q100 assemblies within the >1.78 Mb encompassing the IGH, as no SNVs were found between the two, and only 102 small indels were detected (indel rate of 5.7 per 100 kb). When we mapped the Q100 Chromosome 14 and our ONT IGH assemblies to the custom IGH reference, we observed, as expected, that large portions of the HG002 IGH locus and corresponding genes were deleted relative to the custom IGH reference (Fig. 3A,B). Notably, our assembly of haplotype 2, corresponding to the paternal haplotype, was missing ∼900 kb of the ∼1.4 Mb IGH sequence in the reference, including all IGHD genes.
Incompleteness of HG002 IGH assemblies. (A) Schematic representation of the diploid IGH assembly of HG002 aligned to our custom IGH reference. Haplotype 1 (H1) and haplotype 2 (H2) correspond to the maternal and paternal chromosomes, respectively, as derived from the T2T assemblies. The top track displays the IGH gene regions, and the bottom track shows the alignment intervals of HG002 assemblies as blue boxes, with large deletions indicated by solid red lines. (B) Annotation of HG002 IGH assemblies. Empty cells indicate deletion of a gene, and the color of the cell corresponds to the matched allele sequence from the IMGT database. Sequences with no identical match are denoted as “novel.”
Annotating assemblies and discovery of novel alleles
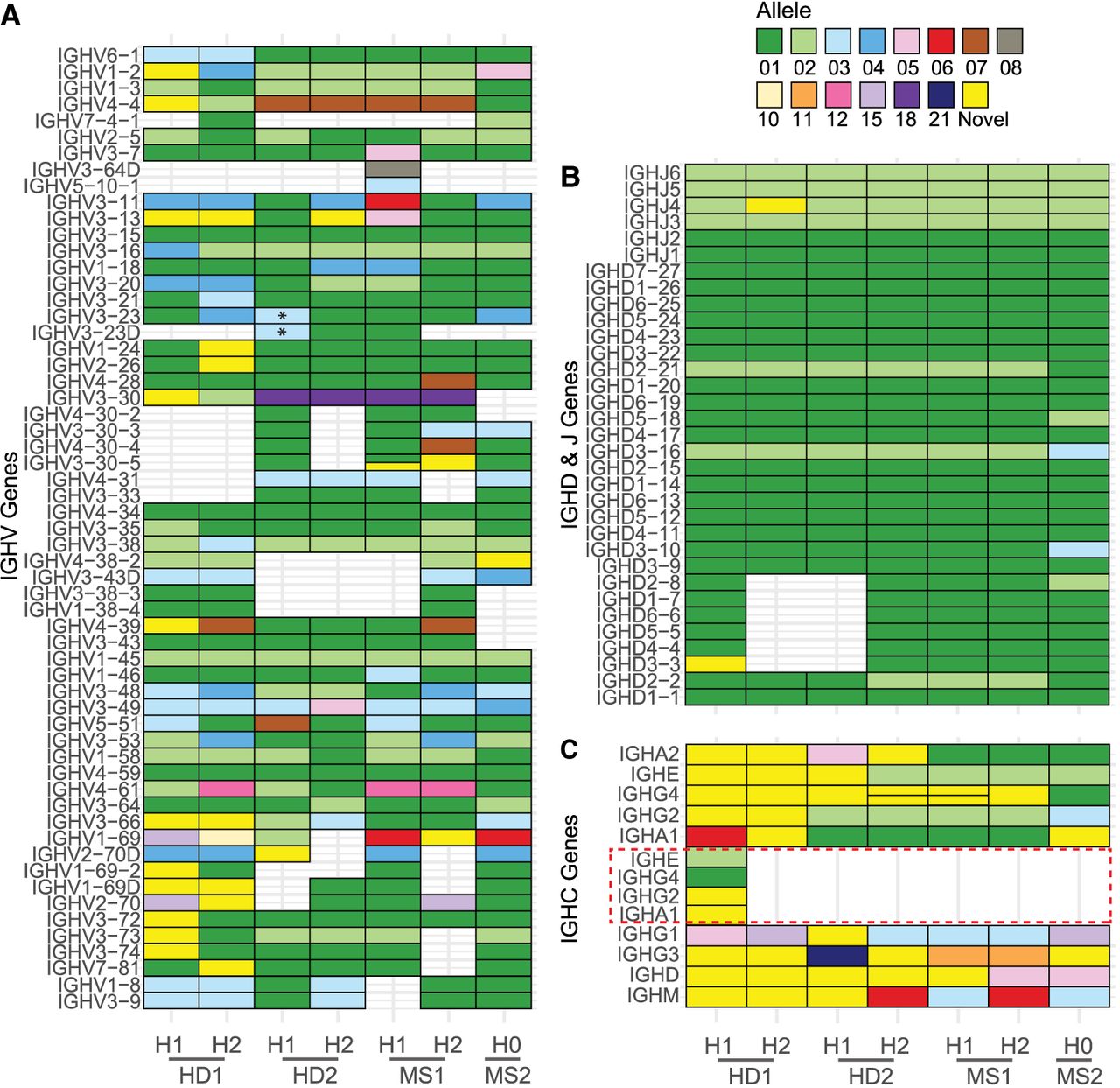
A specialized IGH annotation pipeline was applied to the final haplotype-resolved assemblies. Alleles were assigned according to their match identity to known alleles within the ImMunoGeneTics Information System (IMGT) database. Perfect matches to documented IMGT alleles were assigned their corresponding identifiers, whereas unmatched alleles were classified as “novel” and labeled accordingly. Across the four donor assemblies, we identified a total of 650 V, D, J, and C genes, from which we annotated 219 distinct alleles (Fig. 4A–C; Supplemental Table S1). The majority of these alleles were already characterized and cataloged by IMGT, whereas 21 of the remaining 49 alleles were found in VDJbase (as of 26.08.2024), and 28 were not found in either database. Notably, of the 69 constant genes annotated in our donors, about half (∼50%) lacked corresponding entries in either IMGT or VDJbase.
Annotation of IGH assemblies. Diagrams depicting genes detected in donors’ IGH assemblies. Empty cell indicates deletion of a gene, and a split cell indicates duplication of a gene. The color of the cell corresponds to the matched allele sequence from the IMGT database; sequences with no identical match are denoted as “novel.” Genes are split by groups: V genes (A), D and J genes (B), and C genes (C). In A, a novel structural variant is denoted by an asterisk and represents a large tandem duplication of IGHV3-23 and/or IGHV3-23D, giving a total of seven copies of IGHV3-23. In C, IGHG4 and its duplicate (sometimes termed IGHG4A or IGHG4D) are joined on one row. The red dotted line indicates a duplication extending from IGHE to IGHA1.
Characterization of a novel large structural variant in the constant region
A potential large collapse in the IGH constant region was found in one of the draft haplotype assemblies of HD1 (Table 2). Inspecting alignment of the donor's reads to the draft assembly confirmed an increased mean read depth in the flagged region, supporting the presence of a structural anomaly. Further analysis of reads mapping to this region revealed intra-haplotype SNVs with two distinct variant patterns, neither of which completely matched the initial uncorrected assembly. A deeper inspection, involving the alignment of all donor ONT reads to our custom IGH reference, revealed three distinct variant patterns across a ∼100 kb interval in the IGHC region. This finding was corroborated by mapping all HiFi reads to the IGH reference, confirming the presence of three distinct variant patterns spanning the IGHE–IGHA1 interval (Fig. 5A).
A 120 kb SV in IGHC. (A) Alignment of PacBio HiFi reads from healthy donor 1 (HD1) onto the hg38 reference genome. The top IGV track shows PacBio HiFi reads grouped by origin: centromeric (H1.1) and telomeric (H1.2) side of the duplication in haplotype 1 (H1), and haplotype 2 (H2). The middle track shows a single ultra-long ONT read (>246 kb) from H1 spanning the duplication, with half of it aligning as a supplementary read. The bottom track illustrates constant gene coordinates. (B) Schematic representation of the large duplication involving four constant genes (IGHE–IGHA1) within HD1 H1. (C,D) Gene usage frequency of each subisotype as determined by FLAIRR-seq in HD1. The duplicated genes (C) are stratified by which part of the duplication they originate, except for IGHG4, in which the H2 and H1.1 alleles were identical (ambiguous). (D) Frequency of usage of the nonduplicated genes from each haplotype. (E) Representative phylogenetic trees from clonal families containing both copies of duplicated constant genes. Branch lengths indicate mutational distances between nodes, and node colors denote subisotype and gene location.
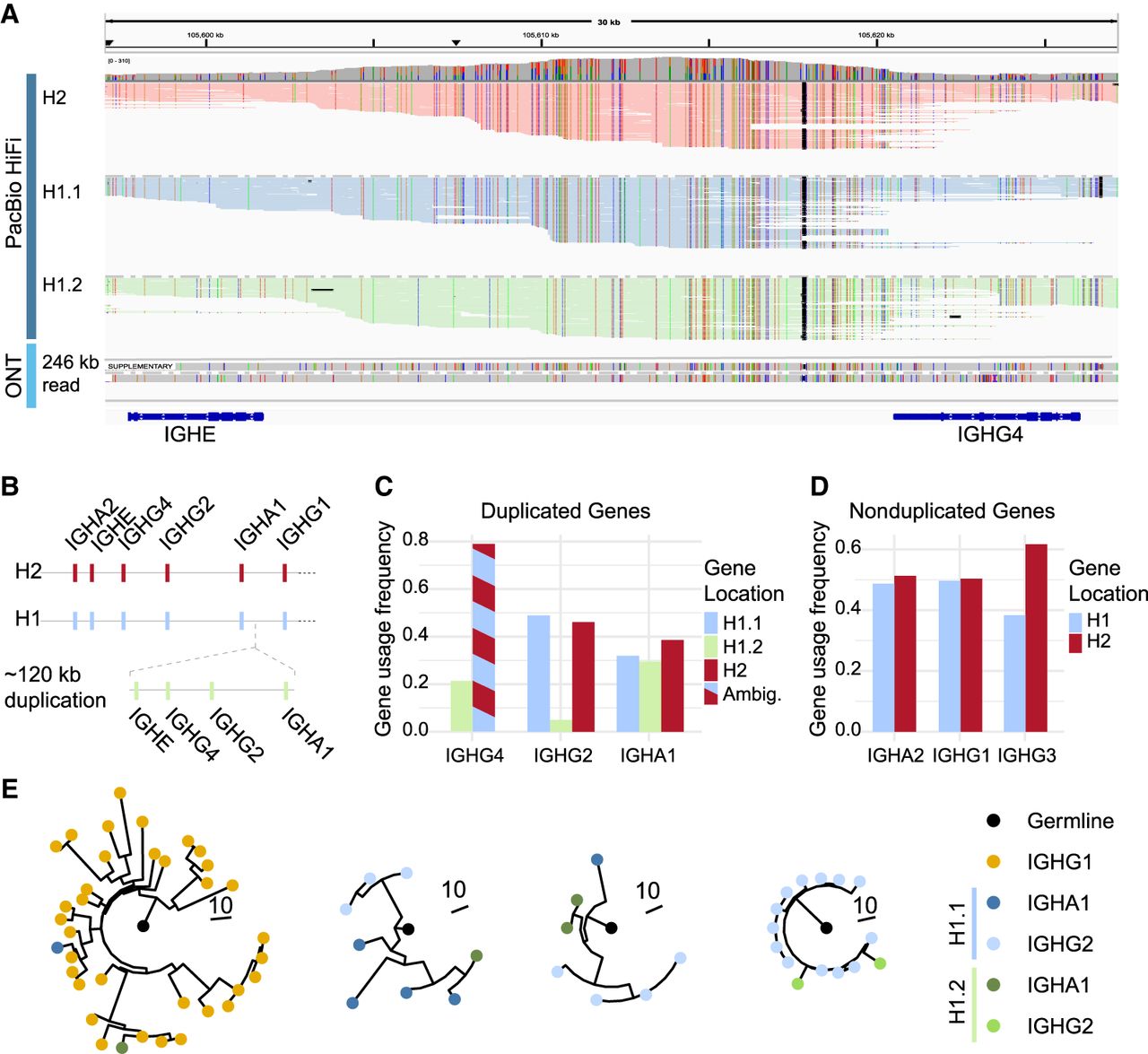
To further dissect this haplotype, we leveraged our ultra-long ONT reads and identified very long reads (>200 kb) that spanned the reference twice over the IGHE–IGHA1 genes. This strongly suggested that two copies of this array were sequentially arranged on the same chromosome. Collectively, these observations pointed to the existence of a large (∼120 kb) duplication in the IGHC region, which had been collapsed during the de novo assembly process. Because of high sequence similarity between the duplicated segments, the variant had to be manually resolved using ultra-long reads spanning the SV, followed by polishing of the assembly with ONT reads. The corrected HD1 haplotype 1 assembly, now including the resolved duplication, showed no conflicts upon realignment of either the ONT or HiFi reads (Table 2).
A proposed model of the SV is presented in Figure 5B. None of the exonic nucleotide sequences of the duplicated genes were identical when compared pairwise. Between the two copies of IGHG2 and IGHG4, there were no coding differences despite three and five exonic SNVs, respectively. IGHA1 contained one nonsynonymous SNV, leading to an amino acid substitution at position 176. The centromeric copy matched the IGHA1*06 allele, whereas the telomeric copy was a novel allele with a D176E mutation. IGHE exhibited the greatest variation among the duplicated genes, with three synonymous and three nonsynonymous SNVs. The telomeric copy corresponded to the IGHE*02 allele, whereas the centromeric copy was a novel allele with mutations A330V, W334G, and L464I. This novel allele most closely resembled IGHE*05 from our reference set but differed from the IMGT allele owing to the A330V and W334G substitutions.
To determine the extent to which both copies of the duplicated genes were expressed in the BCR repertoire, we performed near-full-length adaptive immune receptor repertoire sequencing (FLAIRR-seq) on peripheral blood mononuclear cells (PBMCs) from the donor (Ford et al. 2023). Notably, we identified productive transcripts from both copies of IGHA1 and IGHG2 within the duplicated haplotype 1 (Fig. 5C). Because the centromeric copy of IGHG4 on haplotype 1 is identical to that on haplotype 2, calling the haplotype for IgG4 transcripts was impossible. The calculated frequency of gene copy usage for each subisotype was evenly distributed between the three copies of IGHA1 (ranging from 29% to 38%). In contrast, IGHG2 transcripts were predominantly derived from the centromeric copy of the gene on haplotype 1 (49%) and the gene on haplotype 2 (46%), whereas the telomeric IGHG2 on haplotype 1 accounted for only 4.9% of the IGHG2 transcript pool. The basis of this biased class‐switching remains unknown (Supplemental Text; Supplemental Fig. S2). For nonduplicated constant region genes, transcripts of IGHA2 and IGHG1 were evenly derived from both haplotypes, whereas IGHG3 from haplotype 2 accounted for 61% of the IGHG3 transcripts (Fig. 5D). Finally, we inferred clonal relationships among transcripts from haplotype 1 using SCOPer (Nouri and Kleinstein 2018) and identified numerous trees in which both copies of a duplicated gene were actively utilized within the same clonal family (example trees are shown in Fig. 5E).
Novel structural variants in the variable region
The donors also harbored several significant novel SVs within the IGHV gene region. In HD2, we characterized the first haplotype with seven copies of the IGHV3-23 gene (Fig. 6A), the longest expansion so far identified. CNV has been noted for this region previously (Sasso et al. 1995), including the recent association with IGHV3-23/D frequency in the expressed Ab repertoire (Rodriguez et al. 2023). We extracted the repetitive ∼10.6 kb segments containing IGHV3-23 and compared them to each other (Fig. 6B). The five centromeric segments had identical V gene sequence, corresponding to the IGHV3-23*03 allele. The two telomeric copies also shared identical nucleotide sequence corresponding to the IGHV3-23*01 allele. Notably, this donor had two additional copies of IGHV3-23*01 on the other haplotype, resulting in nine diploid copies of this V gene.
Characterization of a seven-copy IGHV3-23 haplotype. (A) Structural variant giving a seven-copy haplotype of IGHV3-23 in HD2. Illustration shows the order of the seven homologous segments. (B) Synteny plot showing sequence identity and structural variation between the seven consecutive IGHV3-23-containing segments. Similarity between segments is represented by lines colored by the percentage identity of matched bases in 250 bp bins. (C) V gene usage in IgG1 transcripts derived from each haplotype in HD2, a “canonical” two-copy and the novel seven-copy IGHV3-23 haplotype. (D) Frequency of allele usage in IgM transcripts containing IGHV3-23 from the seven-copy haplotype.
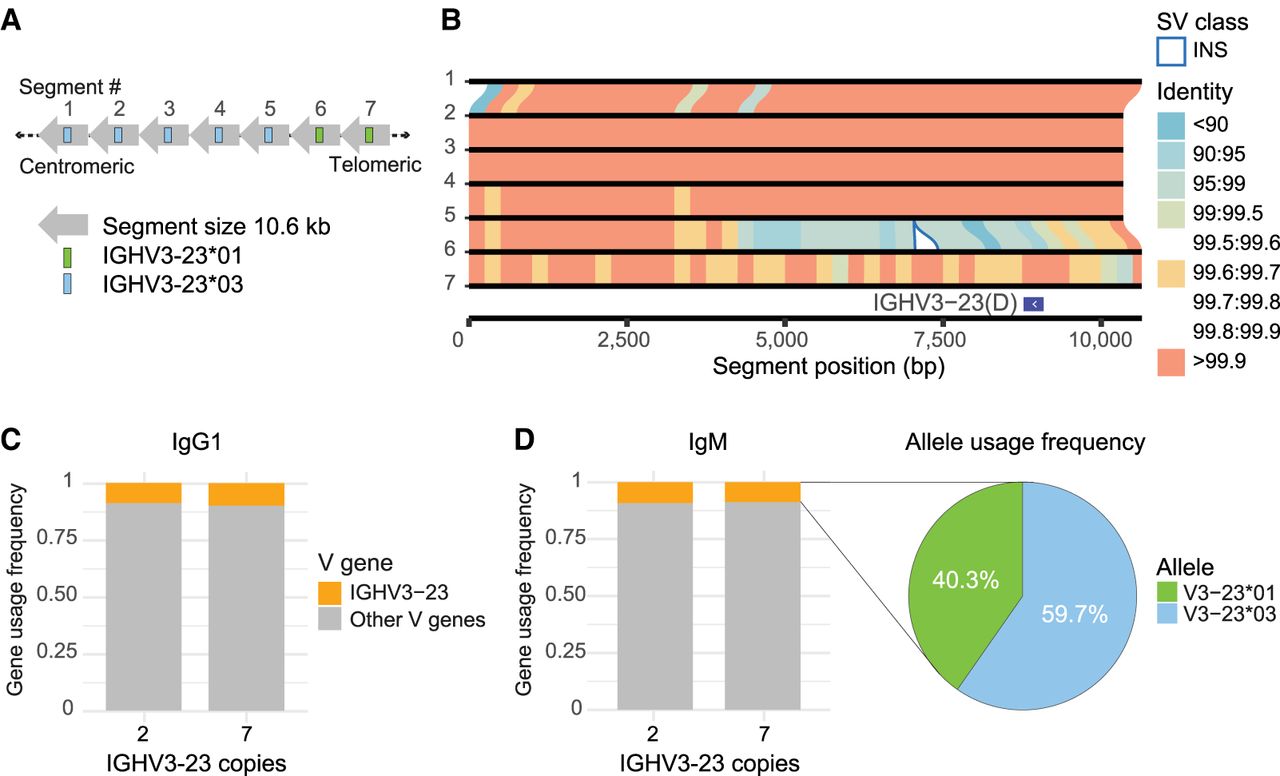
To investigate whether the IGHV3-23 gene expansion translated to proportionally higher usage in the expressed BCR repertoire, we performed FLAIRR-seq on PBMCs from the donor. HD2 is heterozygous for IGHG1 and IGHM, which allowed us to differentiate IgG1 and IgM transcripts derived from the two-copy and the seven-copy IGHV3-23 haplotype. Our analysis revealed no difference in the proportional usage of IGHV3-23 between the haplotypes, in neither the IgG1 nor IgM repertoires (Fig. 6C,D), despite the extra five copies on one haplotype. In the seven-copy haplotype, IgM transcripts utilizing the IGHV3-23 gene were more frequently derived from the *03 allele than the *01 allele (Fig. 6D), aligning with the greater number of *03 allele copies.
In addition, HD2 carried single-copy IGHV1-69/IGHV2-70 haplotypes on each of their chromosomes. However, analysis of these haplotypes in the context of the GRCh38 two-copy IGHV1-69/IGHV2-70 haplotype (which harbors IGHV1-69D, IGHV1-69-2, and IGHV2-70D) indicated that they represent distinct deletion breakpoints. Breakpoint analysis of haplotype 2 was consistent with the deletion of IGHV1-69, IGHV2-70D, and IGHV1-69-2 (Supplemental Fig. S3D). However, determination of breakpoint in haplotype 1 was less conclusive as the haplotype is less congruent with the reference (Supplemental Fig. S3C). The haplotype lacks IGHV1-69-2 and IGHV1-69D, but fine mapping of the breakpoint indicates it occurred close to the boundary of, or within, IGHV2-70D or IGHV2-70. This means the deletion may not technically be a deletion of one or the other gene but rather a hybrid of the two. Critically, this is in contrast to the single-copy IGHV1-69/IGHV2-70 haplotype in MS1, which represents the deletion of IGHV1-69D, IGHV1-69-2, and IGHV2-70D, similar to the gene deletion profile previously characterized in GRCh37 (Rodriguez et al. 2023).
Hemizygous deletion or identical IGH haplotypes
One of the MS patients (MS2) lacked heterozygous variants necessary for accurate phasing of the IGH locus. Consequently, a nonphased single-haplotype assembly was generated from all reads in the locus. Although a small number of heterozygous variants were detected (Supplemental Fig. S4A), closer examination revealed that these variants had low read support and were located in highly repetitive regions of the locus, consistent with known sequencing errors associated with the ONT platform (Sereika et al. 2022). The quality distribution of the few heterozygous variants mirrored this observation, in that most variants were of low quality (Supplemental Fig. S4B). Furthermore, when corresponding HiFi reads were aligned on the haploid assembly, only one candidate SNV was called (Fig. 2A). These findings suggest that this individual either is hemizygous for the IGH locus or possesses two near-identical haplotypes. Additional coverage analysis of HiFi reads from the patient supports the hypothesis of a diploid locus, as no reduction in read depth was seen in IGH compared with other loci, and their coverage pattern mirrored those of the other donors (Supplemental Fig. S5). Accordingly, further bioinformatic separation of the two haplotypes was not warranted.
Discussion
In this study, we present a method that utilizes ONT ultra-long sequencing and adaptive sampling to generate phased, accurate, and complete IGH assemblies without the need for complementary support by other technologies. By employing DNA extraction techniques to preserve ultra-long reads, we achieve an N50 exceeding 65 kb, with some reads extending beyond several hundred kilobases. This is complemented by a bioinformatic framework that assembles, phases, quality controls, and comprehensively annotates all IGH genes. Notably, our method facilitates phasing across larger intervals than comparable methods, consistently resolving the entire IGH locus into single-contig haplotypes. The value of the method is further demonstrated by its ability to uncover significant SV, even in a very limited cohort.
Among these findings is a ∼120 kb duplication in the IGHC region encompassing IGHE, IGHA1, IGHG2, and IGHG4, which to our knowledge is the first sequence-level description of this variant. A duplication in the same region was reported >30 years ago in an Italian family using Southern blot analysis (Bottaro et al. 1993); however, it is not possible to determine whether this duplication corresponds to the same haplotype or represents an individually arising variant. To this point, such large homologous duplications have been impossible to resolve using short-read technology and challenging to discover serologically when relying on sparse coding variation. Importantly, the variant appears to have functional consequences for the expressed B cell receptor repertoire, as the sequencing of IgA and IgG transcripts from the donor showed productive transcripts containing all three copies of IGHA1 and IGHG2 (Fig. 5C). Gene-usage frequencies differed by isotype: 61.5% of IGHA1 transcripts derived from the duplicated haplotype, whereas one IGHG2 copy accounted for <5% of its transcripts. Moreover, we observed clonal families using both duplicated copies within the same lineage (Fig. 5E), suggesting either noncanonical switching between copies (e.g., IGHG4 → IGHA1) or independent switching events in expanding B cell clones.
Another significant discovery was a large tandem duplication expansion of IGHV3-23. This haplotype had seven copies of the IGHV3-23 gene, wherein five consecutive segments contained the *03 allele, and the last two were *01 (Fig. 6A). The five ∼10.6 kb duplicated segments containing the *03 allele displayed almost identical sequences even beyond the V gene exon (Fig. 6B), which suggests that some multicopy IGHV3-23/D haplotypes will require long reads (>50 kb) to correctly resolve CNV. However, designation of these duplication blocks to those harboring IGHV3-23 or IGHV3-23D among the two-copy is nontrivial, suggesting that analysis of additional multicopy haplotypes from diverse populations will likely be required to effectively characterize gene copy number, allelic polymorphisms, and noncoding SNVs.
We were interested in the effect of this expansion on the expressed repertoire, as HD2 carried a total of nine diploid copies of the IGHV3-23 gene. IGHV3-23 is associated with influenza response (Burton et al. 2022) and is one of the most used variable genes in the BCR repertoire (He et al. 2022). It has been shown that CNV of IGHV3-23 has a significant additive effect on its gene usage (Rodriguez et al. 2023). Therefore, it was surprising to find that IGHV3-23 usage fell within the range of what has previously been published and that there was no difference in gene usage between the haplotypes despite a 3.5-fold difference in gene copy number between them (Fig. 6C,D).
Interesting deletion variants were also present in our donor cohort, exemplified by the single-copy IGHV1-69/2-70 gene haplotypes (Supplemental Fig. S3A–D). Closer analysis of putative deletion breakpoints among these haplotypes indicates that this region is likely the site of recurrent SVs and that there are at least two deletion haplotypes circulating in the human population. These deletion haplotypes, in addition to the other SVs identified here, highlight considerations for the characterization and cataloging of both coding and noncoding variation in these regions, in particular as this pertains to the naming and curation of gene allelic variation. For example, with respect to the IGHV1-69/D and IGHV2-70/D genes, alleles from single-copy haplotypes have been historically assigned to IGHV1-69 and IGHV2-70. However, our analysis shows that the accurate allele assignment will require more nuanced and high-resolution analysis of gene copy number and haplotype variation to make such designations. Likewise, it will be nontrivial to name and assign alleles to the new IGHV3-23 and IGHC genes identified in this study and highlights that we should proceed with caution to do this robustly.
We observed a distinctly different variant profile between the donors during phasing, most notably in MS2, which showed a significant skew toward homozygous variants (Supplemental Fig. S4A). Because accurate phasing relies on a balanced distribution of high-quality heterozygous variants, the absence of this leads to unusually small phase sets in this donor, suggesting two nearly identical IGH haplotypes or hemizygosity at the locus. Although terminal 14q deletions have been reported (Maurin et al. 2006; Geier et al. 2017), they are rare and typically accompanied by developmental phenotypes that our donor did not exhibit. Moreover, the donor had normal serum levels of IgA, IgM, and IgG (Supplemental Table S2) and no history of increased susceptibility to infection. Coverage analysis using PacBio HiFi reads at other chromosomes likewise showed no IGH-specific depth reduction (Supplemental Fig. S5). Although techniques such as in situ hybridization or qPCR could have confirmed copy number, these assays were beyond the scope of this study.
It is evident that existing databases are far from covering the full extent of allelic polymorphism in Ig genes within the human population (Gadala-Maria et al. 2015; Rodriguez et al. 2023). In our study of only four individuals, we identified 24 novel IGHV alleles and 23 novel IGHC alleles not present in the IMGT database. These novel alleles were independently validated using mapped HiFi reads, with each allele supported by at least 10 HiFi reads. Compared with allele inference using adaptive immune receptor repertoire sequencing (AIRR-seq), a major advantage of our method is its ability to detect variants in the noncoding regions of IGH. This is in the same vein as the established PacBio SMRT-seq and DNA capture probe method, which was recently used to explore the impact of germline variation on the expressed repertoire (Rodriguez et al. 2023). The study revealed that SNVs associated with variation in IGHV gene usage were enriched in intergenic regions involved in VDJ recombination, thereby explaining previous observations of biased allele usage inferred from AIRR-seq data (Gidoni et al. 2019).
The genetic sequence of different EBV-LCLs have been examined extensively in order to characterize the human genome. However, EBV-LCLs are established by infecting a heterogeneous pool of mature B cells with EBV, resulting in the creation of immortalized cell lines from cells that have undergone recombination of their IG loci. Therefore, depending on the clonality of the transformed B cell pool that makes up the EBV-LCL, substantial portions of the original germline IGH locus are expected to be absent (Rodriguez et al. 2021). This correlates well with our findings of significant deletions in both the maternal and paternal haplotypes of the HG002 EBV-LCL at the IGH locus (Fig. 3A). The clonality of our HG002 cell line is unknown, but as the IGH assemblies resolved without ambiguity and were nearly identical to the T2T-GIAB Q100 assemblies, we should assume clonality is low if not monoclonal. This aligns with a recent paper that found a low number of recombination events in HG002 using data from the Human Pangenome Reference Consortium (Lin et al. 2025).
Our method successfully assembled all IGH haplotypes from four donors and one cell line into single-phase blocks and resolved novel structural variants. To achieve this, we had to optimize several key steps in our pipeline. First, to ensure consistent results across all individuals, we experimented with various assembly and phasing strategies. For instance, we experienced that creating a consensus haploid assembly as a starting point before converting it into a diploid assembly, as suggested by others (Beaulaurier et al. 2025), often led to misassemblies if there were large SVs between the two haplotypes. Consequently, we found that a reference-based phasing strategy produced the most consistent phasing and assemblies for all individuals. Second, an essential component of our pipeline is the quality control of the draft assemblies, which involves mapping reads back to the assembly draft in a haplotype-aware manner. Misassemblies were detected using Flagger, which estimates the expected read coverage and flags regions with coverage inconsistencies that are likely to be assembly errors (Liao et al. 2023). This enabled us to identify issues such as the large duplication in the constant region in HD1, which required manual curation.
The historically low read accuracy of ONT reads has previously posed a challenge for using the technology independently, often necessitating a combination with short-read technologies to enhance accuracy (Wang et al. 2021). However, recent advancements of the ONT platform have substantially improved its sequencing accuracy (Sereika et al. 2022). In our present pipeline, we utilize a single R10.4.1 flow cell in conjunction with the superaccurate basecalling mode, achieving an estimated mean read accuracy exceeding 99% and a diploid read depth of ∼30× over the IGH locus. This translated into ∼100% accurate assemblies, detecting only three potential SNVs in total and 7.2 indels per 100 kb compared with HiFi reads from an established probe-based framework (Rodriguez et al. 2020). Moreover, benchmarking our method using the HG002 reference genome revealed no SNVs and only 24 indels compared with the Q100 published genome (Rautiainen et al. 2023). This level of accuracy, previously unattainable with sequencing on ONT alone, signifies a milestone in long-read sequencing, enabling the generation of highly accurate assemblies without the need for short-read polishing.
This method has several important limitations. First, adaptive sampling relies on matching the first approximately 200 bases of each read to the IGH reference (Loose et al. 2016), so truly novel sequences within that window could in theory be missed. If those initial bases map to the reference, however, the sequencer will continue through the remainder of the molecule, regardless of any novel sequence downstream. Consistent with this, we observed no drop in read depth around the novel variants described here, indicating that the current reference suffices for the individuals in this study. Second, off-target reads occupy pores and reduce yields, which currently limits throughput for large cohorts. Future improvements to the enrichment algorithm could allow multiplexing samples on the same flow cell and simultaneously enrich other immune loci (e.g., IG light chains, TR, HLA), paving the way for a more comprehensive characterization of germline immune genetics. Third, existing phasing and assembly tools are not fully optimized for the complexities of the IGH locus, forcing manual inspection of intermediary files and making the workflow labor intensive. As more IGH haplotypes become available, pan-locus reference graphs and graph-based variant callers may well replace de novo assembly entirely. Finally, compared with the capture-based PacBio protocol (Rodriguez et al. 2020), our ultra-long ONT workflow produces reads ≥100 kb, ideal for resolving large structural variants and achieving full IGH phasing but with a higher cost, a longer turnaround, and greater compute requirements. We currently dedicate one PromethION R10.4.1 flow cell per sample (about $1000 for flow cell + ULK V14 kit), with ∼48 h sequencing plus ∼12 h GPU basecalling on a high-end card (total ∼60 h). In contrast, the PacBio Revio platform enables multiplexing of up to five libraries per SMRT Cell, with each library comprising six samples, effectively allowing 30-plex sequencing. It generates ∼15–20 Gb HiFi data per sample in ∼24 h via on-instrument CCS calling and incurs only standard CPU demand at a consumable cost of about $250–300 per sample. We are currently testing sequential loading of two samples for ultra-long ONT libraries, which should halve consumable costs and help close the time-to-data gap.
In summary, the method described in this paper provides substantial utility for characterizing the IGH locus in both health and disease contexts, a task that has historically been challenging owing to the limitations of short-read sequencing in resolving intricate genetic sequences and structures. This approach opens new avenues for exploring genetic diversity and commonalities across populations and patient cohorts, facilitating the discovery of inherited genetic patterns that influence adaptive immune responses.
Methods
Ethics statement
The study and sample collection was approved by the regional ethical committee of SouthEast Norway (2009/23) and the University of Louisville institutional review board (IRB 14.0661). All donors provided written informed consent before study inclusion.
Sample collection and preparation for ONT sequencing
PBMCs were collected from two MS patients using Vacutainer CPT (Becton Dickinson Biosciences), before monocytes were negatively selected using the Pan Monocyte Isolation Kit, human (Miltenyi Biotec). Cells were frozen and kept in nitrogen until DNA extraction and sequencing as stated below.
Commercial frozen human PBMCs were purchased from StemCell Technologies. HG002 EBV-LCL were purchased from the Coriell Institute of Medical Research and expanded for 14 days in Gibco RPMI 1640 (Thermo Fisher Scientific) supplemented with 15% heat-inactivated fetal calf serum and penicillin/streptomycin at 100 U/mL and 100 µg/mL, respectively, before aliquots were freezed.
ONT DNA library preparation
Approximately 6 × 106 cells, either monocytes (MS1 and MS2), PBMCs (HD1 and HD2), or EBV-LCLs (HG002), were used as input for DNA extraction and subsequent ultra-long sequencing library preparation. DNA was extracted using Monarch HMW DNA Extraction from Cell & Blood (New England Biolabs) following the protocol “High Molecular Weight DNA (HMW DNA) Extraction from Cells” (NEB T3050) with the following alterations: (1) the agitation speed during cell lysis was set to 1400 rpm for 10 min; (2) the incubation time with beads during gDNA binding to beads was increased to 8 min; (3) the elution buffer was replaced with 560 µL extraction EB from SQK-ULK114 (ONT); (4) an extra incubation step for 1 h at room temperature was added after elution with agitation at 56°C; (5) the centrifugation of beads with DNA was increased to 1 min at 16,000g; and (6) an additional 200 µL extraction EB buffer was added after complete elution to get correct input volume for ONT library preparation. Sequencing libraries were generated using an Ultra-Long DNA Sequencing Kit from ONT (SQK-ULK114) following the manufacturer's guidelines. In line with recent literature and ONT documentation, we use the term “ultra-long” for libraries with N50 > 50 kb that also include a substantial proportion of bases in reads >100 kb.
ONT sequencing
Sequencing was performed on a PromethION 2 solo (P2 solo) device connected to a high-performance computer running MinKNOW software version 23.07.12. The loading and washing of PromethION R10.4.1 flow cells were performed as per the manufacturer's guidelines. Each donor was sequenced using one flow cell, and the sequencing ran for a maximum of 72 h with washing and reloading performed around every 24 h using the Flow Cell Wash Kit (EXP-WSH004) to increase output.
To serve as an IGH reference for adaptive sampling during sequencing, T2T-CHM13v2.0 was altered at the telomere end of Chromosome 14. Here, the variable part of IGH was replaced with a custom IGH reference as described elsewhere (Rodriguez et al. 2020), whereas the rest of Chr 14 including the IGH constant region remained. Adaptive sampling was enabled in MinKNOW to enrich for the ROI, specified as the IGH region with an additional 200 kb lead-in from the custom CHM13 reference. During sequencing, the high accuracy basecalling model was enabled to accommodate adaptive sampling. The resulting POD5 files were rebasecalled post-run using the superaccurate model, filtering out reads with a Phred score below 10, and were subsequently used for further downstream processing.
Processing ONT data to IGH assemblies
A pipeline to process ONT reads into haplotype-resolved IGH assemblies was established using existing tools and custom scripts. First, catfishq (version 1.4.0) (https://github.com/philres/catfishq) was used to filter out adaptive sampling rejected reads from the rebasecalled FASTQ files. Next, the reads were aligned to our CHM13v2.0 reference with the custom IGH described above using minimap2 (v 2.26) with the option “-ax map-ont” (Li 2018). Variant calling was performed on the resulting BAM file with Clair3 (v 1.0.0) (Zheng et al. 2022) with the options “‐‐enable_phasing”, ‘‐‐platform=’ont”, and “‐‐bed_fn” enabled, specifying a BED file with IGH coordinates (chr14:99630000-101325184). Reads were haplotagged using Clair3 <phased_merged_output.vcf> with WhatsHap (v 1.4) (Martin et al. 2016) haplotag and the options “‐‐output-haplotag-list ‐‐ignore-read-groups ‐‐tag-supplementary ‐‐region=chr14:99630000-101325184”. Continuity of haplotype tags between phase sets was evaluated by manual inspection of the haplotagged BAM in the Integrative Genomics Viewer (IGV) (Robinson et al. 2017). If switch errors were detected, the Clair3 VCF was edited before running “whatshap split” with default parameters to produce hap1 and hap2 phased FASTQs. Lastly, de novo assembly was produced from the haplotype-separated reads using Flye (v 2.9.2) (Kolmogorov et al. 2019) with the arguments “‐‐genome-size 1200K ‐‐nano-hq <phased.fastq>”.
Manual inspection in IGV and Flagger (v 0.4.0) (Liao et al. 2023) was used to evaluate haplotype-resolved assemblies. ONT reads from the ROI were mapped back to the diploid assembly with minimap2- and Flagger-highlighted misassembled regions. Problematic regions of the assemblies had to be manually resolved when collapsed by the assembler. In the example of the collapsed large duplication in the IGHC region, we isolated reads that spanned the duplication. Subsequently, one of the longer isolated reads was extracted to a FASTA and polished with the rest of the isolated reads using “flye ‐‐nano-hq <isolated_reads.fastq> ‐‐polish-target <long_read.fasta> ‐‐iterations 3”. The polished long read was mapped back to the first-draft assembly with minimap2 with the “-ax asm20” option, and coordinates for cutting and pasting of the resolved duplication were determined. The entire collapsed sequence was removed, and the polished long read was trimmed to fit this gap in the assembly and inserted here. The resulting new draft assembly was finally verified again as outlined above with Flagger.
PacBio HiFi sequencing of IGH using capture probes
Parallel sequencing of samples from all donors was performed according to a previously published method (Rodriguez et al. 2020). In brief, probe-based targeted capture of the IGH region and long-read single-molecule, real-time (SMRT)-seq was used to sequence IGH reads. Libraries were sequenced on PacBio instruments SEQUEL II (HD1, MS1, and MS2) or REVIO (HD2).
IgA and IgG FLAIRR-seq
RNA was extracted from HD1 and HD2 PBMCs using the AllPrep DNA/RNA Mini Kit (Qiagen). IgG and IgA transcripts were resolved by targeted amplification using FLAIRR-seq as described previously (Ford et al. 2023). FLAIRR-seq cDNA libraries were prepared using the SMRTbell prep kit 3.0 (PacBio) according to the manufacturer's specifications. The prepared library was sequenced on the Revio long-read system (PacBio). FLAIRR-seq data were processed via the Immcantation tool suite using pRESTO and Change-O as previously described (Vander Heiden et al. 2014; Gupta et al. 2015; Ford et al. 2023). For the identification of clonal relationships, we used SCOPer hierarchical clustering with a threshold of 0.15 (Gupta et al. 2017). Dowser and Igphyml were used to build and visualize the clonal trees (Hoehn et al. 2019, 2022).
Validation of IGH assemblies
Personalized alignment of HiFi long reads were used to infer orthogonal validation, and base-level concordance was obtained to ensure the highest quality of haplotype-resolved assemblies. The HiFi reads were aligned with minimap2 to the corresponding ONT IGH assemblies with the option “-ax map-hifi”. The variant caller Clair3 was used to assess base accuracy as a measure of agreement between the high-accuracy reads and the polished assemblies. Clair3 image was ran with singularity and the options “‐‐platform=‘hifi’ ‐‐model_path=‘hifi_sequel_2’ ‐‐no_phasing_for_fa”.
The alignment of HiFi reads were also used to infer orthogonal validation, and base-level concordance was obtained with other metrics. Custom Python scripts were utilized to assess the proportion of assembly bases with at least 80% concordance from the reads, ensuring a minimum read depth of 5× and 10× for estimating coverage and accuracy.
Annotating IGH assemblies
IGH assemblies were annotated in a reference-guided approach using custom Python and bash scripts available at GitHub (https://github.com/Watson-IG/wasp) and as Supplemental Code. Finished ONT assemblies and reads were aligned to a human reference genome (Rodriguez et al. 2020) using minimap2 with the presets “-ax asm20” and “-ax map-ont”, respectively. Sequences for IGH exons were extracted from assembly BAM using gene coordinates mapped to the reference from GRCh38 and searched against available human IGH alleles in IMGT database (IGHV, -D and -J sets were downloaded February 13, 2024; IGHC alleles were collected November 24, 2022). HiFi reads mapping over the same intervals were used to validate support for assigned and novel alleles, using the same Python scripts and metrics that were used to assess read coverage in the assembly as a whole.
Data access
The sequencing data for HG002 generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1196145. Sequencing data and complete IGH assemblies from samples HD1, HD2, MS1, and MS2 generated in this study have been submitted to the European Genome-phenome Archive (EGA; https://ega-archive.org) under accession numbers EGAS50000001042 and EGAD50000001517.
Competing interest statement
C.T.W., M.L.S., and W.D.L. are founders and shareholders of Clareo Biosciences and serve on its executive board. M.L.S. and C.T.W. are listed inventors of patent-filing PCT/US2024/044692. None of the other authors declare any competing interests.
Acknowledgments
We thank the patients and donors whose contributions were vital to this study. We thank Steffan Daniel Bos-Haugen for useful discussions in the initiation of this project, Milda Kaniušaitė for her valuable guidance on the wet laboratory and sequencing protocol, and Professor Tone Tønjum for providing access to sequencing equipment during the initial phase of the study. This research was supported by grants from the Research Council of Norway (project no. 314376; A.L.) and the Southeastern Norway Regional Health Authority (project no. 2021085; A.L.). Additional support, in part, was provided by the National Institute of Allergy and Infectious Diseases (R24 AI138963; C.T.W. and M.L.S.). SMRT sequencing on the PacBio Revio system was enabled by National Institutes of Health instrumentation award S10 OD034432 (M.L.S.).
Author contributions: A.L., E.R., and C.T.W. contributed to the conception of the work. M.B.G., E.R., and E.E.F. performed the sequencing. M.B.G., A.L., A.T., W.D.L., Z.V., U.J., and C.T.W. developed the computational pipeline to analyze the ONT sequencing data. M.B.G., A.L., U.J., A.T., C.T.W., and E.E.F. analyzed sequencing data from ONT, PacBio, and/or FLAIRR-seq. A.L., E.R., M.L.S., and C.T.W. acquired funding. M.B.G. and A.L. drafted the manuscript. All authors contributed to revising the manuscript.
Notes
[2] Supplementary material [Supplemental material is available for this article.]
[3] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280400.125.
References
- ↵Avnir Y, Watson CT, Glanville J, Peterson EC, Tallarico AS, Bennett AS, Qin K, Fu Y, Huang C-Y, Beigel JH, 2016. IGHV1-69 polymorphism modulates anti-influenza antibody repertoires, correlates with IGHV utilization shifts and varies by ethnicity. Sci Rep 6: 20842. 10.1038/srep20842
- ↵Bashirova AA, Zheng W, Akdag M, Augusto DG, Vince N, Dong KL, O'hUigin C, Carrington M. 2021. Population-specific diversity of the immunoglobulin constant heavy G chain (IGHG) genes. Genes Immun 22: 327–334. 10.1038/s41435-021-00156-2
- ↵Beaulaurier J, Ly L, Duty JA, Tyer C, Stevens C, Hung C-T, Sookdeo A, Drong AW, Kowdle S, Guzman-Solis A, 2025. De novo antibody identification in human blood from full-length single B cell transcriptomics and matching haplotype-resolved germline assemblies. Genome Res 35: 929–941. 10.1101/gr.279392.124
- ↵Bottaro A, Gallina R, Brusco A, Cariota U, Boccazzi C, Barilaro MR, Plebani A, Ugazio AG, van Leeuwen AM, DeLange GG, 1993. Familial clustering of IGHC deletions and duplications: functional and molecular analysis. Immunogenetics 37: 356–363. 10.1007/BF00216800
- ↵Buck D, Albrecht E, Aslam M, Goris A, Hauenstein N, Jochim A, Cepok S, Grummel V, Dubois B, Berthele A, 2013. Genetic variants in the immunoglobulin heavy chain locus are associated with the IgG index in multiple sclerosis. Ann Neurol 73: 86–94. 10.1002/ana.23749
- ↵Burton AR, Guillaume SM, Foster WS, Wheatley AK, Hill DL, Carr EJ, Linterman MA. 2022. The memory B cell response to influenza vaccination is impaired in older persons. Cell Rep 41: 111613. 10.1016/j.celrep.2022.111613
- ↵Calonga-Solís V, Malheiros D, Beltrame MH, Vargas LDB, Dourado RM, Issler HC, Wassem R, Petzl-Erler ML, Augusto DG. 2019. Unveiling the diversity of immunoglobulin heavy constant gamma (IGHG) gene segments in Brazilian populations reveals 28 novel alleles and evidence of gene conversion and natural selection. Front Immunol 10: 1161. 10.3389/fimmu.2019.01161
- ↵Chen X, Sun X, Yang W, Yang B, Zhao X, Chen S, He L, Chen H, Yang C, Xiao L, 2018. An autoimmune disease variant of IgG1 modulates B cell activation and differentiation. Science 362: 700–705. 10.1126/science.aap9310
- ↵Cyster JG, Allen CDC. 2019. B cell responses: cell interaction dynamics and decisions. Cell 177: 524–540. 10.1016/j.cell.2019.03.016
- ↵deCamp AC, Corcoran MM, Fulp WJ, Willis JR, Cottrell CA, Bader DLV, Kalyuzhniy O, Leggat DJ, Cohen KW, Hyrien O, 2024. Human immunoglobulin gene allelic variation impacts germline-targeting vaccine priming. NPJ Vaccines 9: 58. 10.1038/s41541-024-00811-5
- ↵De Taeye SW, Bentlage AEH, Mebius MM, Meesters JI, Lissenberg-Thunnissen S, Falck D, Sénard T, Salehi N, Wuhrer M, Schuurman J, 2020. Fcγr binding and ADCC activity of human IgG allotypes. Front Immunol 11: 740. 10.3389/fimmu.2020.00740
- ↵Feeney AJ. 1992. Comparison of junctional diversity in the neonatal and adult immunoglobulin repertoires. Int Rev Immunol 8: 113–122. 10.3109/08830189209055567
- ↵Ford EE, Tieri D, Rodriguez OL, Francoeur NJ, Soto J, Kos JT, Peres A, Gibson WS, Silver CA, Deikus G, 2023. FLAIRR-seq: a method for single-molecule resolution of near full-length antibody H chain repertoires. J Immunol 210: 1607–1619. 10.4049/jimmunol.2200825
- ↵Gadala-Maria D, Yaari G, Uduman M, Kleinstein SH. 2015. Automated analysis of high-throughput B-cell sequencing data reveals a high frequency of novel immunoglobulin V gene segment alleles. Proc Natl Acad Sci 112: E862–E870. 10.1073/pnas.1417683112
- ↵Geier CB, Piller A, Eibl MM, Ciznar P, Ilencikova D, Wolf HM. 2017. Terminal 14q32.33 deletion as a novel cause of agammaglobulinemia. Clin Immunol 183: 41–45. 10.1016/j.clim.2017.07.003
- ↵Gidoni M, Snir O, Peres A, Polak P, Lindeman I, Mikocziova I, Sarna VK, Lundin KEA, Clouser C, Vigneault F, 2019. Mosaic deletion patterns of the human antibody heavy chain gene locus shown by Bayesian haplotyping. Nat Commun 10: 628. 10.1038/s41467-019-08489-3
- ↵Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, Kleinstein SH. 2015. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31: 3356–3358. 10.1093/bioinformatics/btv359
- ↵Gupta NT, Adams KD, Briggs AW, Timberlake SC, Vigneault F, Kleinstein SH. 2017. Hierarchical clustering can identify B cell clones with high confidence in Ig repertoire sequencing data. J Immunol 198: 2489–2499. 10.4049/jimmunol.1601850
- ↵He B, Liu S, Xu M, Hu Y, Lv K, Wang Y, Ma Y, Zhai Y, Yue X, Liu L, 2022. Comparative global B cell receptor repertoire difference induced by SARS-CoV-2 infection or vaccination via single-cell V(D)J sequencing. Emerg Microbes Infect 11: 2007–2020. 10.1080/22221751.2022.2105261
- ↵Hoehn KB, Vander Heiden JA, Zhou JQ, Lunter G, Pybus OG, Kleinstein SH. 2019. Repertoire-wide phylogenetic models of B cell molecular evolution reveal evolutionary signatures of aging and vaccination. Proc Natl Acad Sci 116: 22664–22672. 10.1073/pnas.1906020116
- ↵Hoehn KB, Pybus OG, Kleinstein SH. 2022. Phylogenetic analysis of migration, differentiation, and class switching in B cells. PLoS Comput Biol 18: e1009885. 10.1371/journal.pcbi.1009885
- ↵Kolmogorov M, Yuan J, Lin Y, Pevzner PA. 2019. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37: 540–546. 10.1038/s41587-019-0072-8
- ↵Lefranc M-P, Lefranc G. 2001. The immunoglobulin FactsBook. Academic Press, London.
- ↵Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191
- ↵Liao W-W, Asri M, Ebler J, Doerr D, Haukness M, Hickey G, Lu S, Lucas JK, Monlong J, Abel HJ, 2023. A draft human pangenome reference. Nature 617: 312–324. 10.1038/s41586-023-05896-x
- ↵Lin M-J, Langmead B, Safonova Y. 2025. IGLoo enables comprehensive analysis and assembly of immunoglobulin heavy-chain loci in lymphoblastoid cell lines using PacBio high-fidelity reads. Cell Rep Methods 5: 101033. 10.1016/j.crmeth.2025.101033
- ↵Loose M, Malla S, Stout M. 2016. Real-time selective sequencing using nanopore technology. Nat Methods 13: 751–754. 10.1038/nmeth.3930
- ↵Lossius A, Tomescu-Baciu A, Holmøy T, Vedeler CA, Røsjø E, Lorentzen ÅR, Casetta I, Vartdal F. 2017. Selective intrathecal enrichment of G1m1-positive B cells in multiple sclerosis. Ann Clin Transl Neurol 4: 756–761. 10.1002/acn3.451
- ↵Martin M, Patterson M, Garg S, Fischer SO, Pisanti N, Klau GW, Schöenhuth A, Marschall T. 2016. WhatsHap: fast and accurate read-based phasing. bioRxiv 10.1101/085050
- ↵Maurin M, Brisset S, Le Lorc'h M, Poncet V, Trioche P, Aboura A, Labrune P, Tachdjian G. 2006. Terminal 14q32.33 deletion: genotype–phenotype correlation. Am J Med Genet A 140A: 2324–2329. 10.1002/ajmg.a.31438
- ↵Mikocziova I, Greiff V, Sollid LM. 2021. Immunoglobulin germline gene variation and its impact on human disease. Genes Immun 22: 205–217. 10.1038/s41435-021-00145-5
- ↵Ni Y, Liu X, Simeneh ZM, Yang M, Li R. 2023. Benchmarking of nanopore R10.4 and R9.4.1 flow cells in single-cell whole-genome amplification and whole-genome shotgun sequencing. Comput Struct Biotechnol J 21: 2352–2364. 10.1016/j.csbj.2023.03.038
- ↵Nouri N, Kleinstein SH. 2018. A spectral clustering-based method for identifying clones from high-throughput B cell repertoire sequencing data. Bioinformatics 34: i341–i349. 10.1093/bioinformatics/bty235
- ↵Rautiainen M, Nurk S, Walenz BP, Logsdon GA, Porubsky D, Rhie A, Eichler EE, Phillippy AM, Koren S. 2023. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat Biotechnol 41: 1474–1482. 10.1038/s41587-023-01662-6
- ↵Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A, Mesirov JP. 2017. Variant review with the Integrative Genomics Viewer. Cancer Res 77: e31–e34. 10.1158/0008-5472.CAN-17-0337
- ↵Rodriguez OL, Gibson WS, Parks T, Emery M, Powell J, Strahl M, Deikus G, Auckland K, Eichler EE, Marasco WA, 2020. A novel framework for characterizing genomic haplotype diversity in the human immunoglobulin heavy chain locus. Front Immunol 11: 2136. 10.3389/fimmu.2020.02136
- ↵Rodriguez OL, Sharp AJ, Watson CT. 2021. Limitations of lymphoblastoid cell lines for establishing genetic reference datasets in the immunoglobulin loci. PLoS One 16: e0261374. 10.1371/journal.pone.0261374
- ↵Rodriguez OL, Safonova Y, Silver CA, Shields K, Gibson WS, Kos JT, Tieri D, Ke H, Jackson KJL, Boyd SD, 2023. Genetic variation in the immunoglobulin heavy chain locus shapes the human antibody repertoire. Nat Commun 14: 4419. 10.1038/s41467-023-40070-x
- ↵Roldán E, Fuxa M, Chong W, Martinez D, Novatchkova M, Busslinger M, Skok JA. 2005. Locus “decontraction” and centromeric recruitment contribute to allelic exclusion of the immunoglobulin heavy-chain gene. Nat Immunol 6: 31–41. 10.1038/ni1150
- ↵Rubelt F, Bolen CR, McGuire HM, Vander Heiden JA, Gadala-Maria D, Levin M, Euskirchen GM, Mamedov MR, Swan GE, Dekker CL, 2016. Individual heritable differences result in unique cell lymphocyte receptor repertoires of naïve and antigen-experienced cells. Nat Commun 7: 11112. 10.1038/ncomms11112
- ↵Sasso EH, Buckner JH, Suzuki LA. 1995. Ethnic differences in VH gene polymorphism. Ann N Y Acad Sci 764: 72–73. 10.1111/j.1749-6632.1995.tb55808.x
- ↵Scharf L, West AP, Gao H, Lee T, Scheid JF, Nussenzweig MC, Bjorkman PJ, Diskin R. 2013. Structural basis for HIV-1 gp120 recognition by a germ-line version of a broadly neutralizing antibody. Proc Natl Acad Sci 110: 6049–6054. 10.1073/pnas.1303682110
- ↵Sereika M, Kirkegaard RH, Karst SM, Michaelsen TY, Sørensen EA, Wollenberg RD, Albertsen M. 2022. Oxford nanopore R10.4 long-read sequencing enables the generation of near-finished bacterial genomes from pure cultures and metagenomes without short-read or reference polishing. Nat Methods 19: 823–826. 10.1038/s41592-022-01539-7
- ↵Tan J, Sack BK, Oyen D, Zenklusen I, Piccoli L, Barbieri S, Foglierini M, Fregni CS, Marcandalli J, Jongo S, 2018. A public antibody lineage that potently inhibits malaria infection through dual binding to the circumsporozoite protein. Nat Med 24: 401–407. 10.1038/nm.4513
- ↵Ternant D, Arnoult C, Pugnière M, Dhommée C, Drocourt D, Perouzel E, Passot C, Baroukh N, Mulleman D, Tiraby G, 2016. IgG1 allotypes influence the pharmacokinetics of therapeutic monoclonal antibodies through FcRn binding. J Immunol 196: 607–613. 10.4049/jimmunol.1501780
- ↵Vander Heiden JA, Yaari G, Uduman M, Stern JNH, O'Connor KC, Hafler DA, Vigneault F, Kleinstein SH. 2014. pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30: 1930–1932. 10.1093/bioinformatics/btu138
- ↵Wang C, Liu Y, Cavanagh MM, Le Saux S, Qi Q, Roskin KM, Looney TJ, Lee J-Y, Dixit V, Dekker CL, 2015. B-cell repertoire responses to varicella-zoster vaccination in human identical twins. Proc Natl Acad Sci 112: 500–505. 10.1073/pnas.1415875112
- ↵Wang Y, Zhao Y, Bollas A, Wang Y, Au KF. 2021. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol 39: 1348–1365. 10.1038/s41587-021-01108-x
- ↵Watson CT, Breden F. 2012. The immunoglobulin heavy chain locus: genetic variation, missing data, and implications for human disease. Genes Immun 13: 363–373. 10.1038/gene.2012.12
- ↵Watson CT, Steinberg KM, Graves TA, Warren RL, Malig M, Schein J, Wilson RK, Holt RA, Eichler EE, Breden F. 2015. Sequencing of the human IG light chain loci from a hydatidiform mole BAC library reveals locus-specific signatures of genetic diversity. Genes Immun 16: 24–34. 10.1038/gene.2014.56
- ↵Yacoob C, Pancera M, Vigdorovich V, Oliver BG, Glenn JA, Feng J, Sather DN, McGuire AT, Stamatatos L. 2016. Differences in allelic frequency and CDRH3 region limit the engagement of HIV Env immunogens by putative VRC01 neutralizing antibody precursors. Cell Rep 17: 1560–1570. 10.1016/j.celrep.2016.10.017
- ↵Yeung YA, Foletti D, Deng X, Abdiche Y, Strop P, Glanville J, Pitts S, Lindquist K, Sundar PD, Sirota M, 2016. Germline-encoded neutralization of a Staphylococcus aureus virulence factor by the human antibody repertoire. Nat Commun 7: 13376. 10.1038/ncomms13376
- ↵Zheng Z, Li S, Su J, Leung AW-S, Lam T-W, Luo R. 2022. Symphonizing pileup and full-alignment for deep learning-based long-read variant calling. Nat Comput Sci 2: 797–803. 10.1038/s43588-022-00387-x