Abstract
Transcription factors (TFs) regulate gene expression by facilitating or disrupting the formation of transcription initiation machinery at particular genomic loci. Because TF occupancy is driven in part by recognition of DNA sequence, genetic variation can influence TF–DNA associations and gene regulation. To identify variants that impact TF binding in human brain tissues, we assessed allele-specific binding (ASB) at heterozygous variants for 94 TFs in nine brain regions from two donors. Leveraging graph genomes constructed from phased genomic sequence data, we compared ChIP-seq signals between alleles at heterozygous variants within each brain region and identified thousands of variants exhibiting ASB for at least one TF. ASB reproducibility was measured by comparisons between independent experiments both within and between donors. We found that rare alleles in the general population more frequently led to reduced TF binding, whereas common alleles had an equal likelihood of increasing or decreasing binding. Further, for ASB variants in predicted binding motifs, the favored allele tended to be the one with the stronger expected motif match, but this concordance was not observed within highly occupied sites. We also found that neuron-specific cis-regulatory elements (cCREs), in contrast with oligodendrocyte-specific cCREs, showed depletion of ASB variants. We identified 2670 ASB variants associated with evidence for allele-specific gene expression in the brain from GTEx data and observed increasing eQTL effect direction concordance as ASB significance increases. These results provide a valuable and unique resource for mechanistic analysis of cis-regulatory variation in human brain tissue.
Gene expression changes occur in essentially every biological process, including the development of diseases such as neurodegenerative (Bonham et al. 2019; Zhao 2023) and psychiatric conditions (Mimmack et al. 2002; Clifton et al. 2019; Huang et al. 2020). Transcription factors (TFs) and their association with DNA are crucial determinants of gene expression, so identifying elements that influence the association between TFs and DNA is key to understanding variation in gene expression. A wide variety of tools have been developed to identify and catalog DNA sequence motifs to which TFs preferentially bind (Bailey et al. 2015; Ghandi et al. 2016; Castro-Mondragon et al. 2022). Although informative, these approaches are limited by the fact that a motif's presence is neither necessary nor sufficient for TF association (Dror et al. 2015), so the impact of DNA sequence changes on motifs is of limited predictive value.
An alternative approach is to assay TF binding behavior at sites of heterozygous variation within an individual tissue donor so as to identify “allele-specific binding” (ASB) as binding sites with a statistically significant excess of reads from one of the two underlying alleles. One issue that confounds detection of ASB is that read alignment methods tend to favor the reference allele, which can increase noise in studies of ASB (Degner et al. 2009; Rozowsky et al. 2011; Smith et al. 2013; Stevenson et al. 2013; Hach et al. 2014). The use of graph structures to represent personalized genomes or pangenomes (Smith et al. 2013; Paten et al. 2017) can reduce reference allele bias (Garrison et al. 2018; Martiniano et al. 2020; Chen et al. 2021).
A recent study probed ASB across hundreds of cell types with corrections for reference allele bias (Abramov et al. 2021). The majority of these data sets were derived from cancer cell lines, limiting their applicability to nondiseased human tissue, or in contexts relevant for specific disease states. Another recent study highlighted the viability of a similar approach in human tissue samples by identifying allele-specific loci in 15 assays in four human donors across 30 tissues, including ChIP-seq assays of histone marks and several TFs, including CTCF (Rozowsky et al. 2011). This study found relationships between allele specificity of ChIP-seq and gene expression, including GTEx eQTLs that exhibited ASB and those that did not. This highlights the value of identifying ASB among TFs for understanding genetic variation of gene regulation.
Here, we greatly expand upon previous work by performing ASB analysis for 920 (Loupe et al. 2024) TF ChIP-seq data sets, spanning 94 distinct TFs in tissue samples from nine anatomically defined brain regions sampled from two donors. For simplicity, in this study we use “TFs” to refer to a broad range of DNA-associated proteins, including transcriptional cofactors and chromatin remodelers, that span a variety of molecular mechanisms and modes of action (Lambert et al. 2018; Loupe et al. 2024). We corrected reference allele bias using an approach combining personalized genomes with WASP, a method that removes reads exhibiting allelic alignment bias (van de Geijn et al. 2015), and found that this approach improves the calling of ASB. We explored the effects of rare genetic variation on the degree of allele specificity and investigated the relationship between ASB and the alteration of TF binding motifs. We measured the enrichment of ASB for several neurological diseases. We determined the effects of ASB on gene expression using RNA-seq from our donors, as well as GTEx eQTLs from the brain, allowing a mechanistic exploration of allele-specific gene regulation. Finally, we highlight an interesting example of ASB identified in our data sets.
Results
Graph genomes improve read mapping and reduce reference allele bias
To study the impact of genetic variation on TF binding, we first performed linked-read sequencing (10x Genomics) to generate phased genomes and call variants for two donors (see Methods) (Fig. 1A; Supplemental Table S1). We built personalized graph genomes, using the vg toolkit (Garrison et al. 2018) to correct for allele-biased mapping, as it has been shown that it can be used for detection of missing signal in histones (Groza et al. 2020) and the detection of allele-biased TF footprints (Ouyang and Boyle 2022). To measure the effectiveness of this approach on our data sets, we mapped a pilot set of 20 ChIP-seq data sets, 10 in each of the two donors, using both conventional mapping to the GRCh38 linear reference via Bowtie 2 and personalized graph-genome mapping via vg. For this analysis, we identified 21,207 heterozygous sites with significant TF–allele bias at a nominal P < 0.05 threshold using GRCh38 (binomial test), 76.9% of which favor the reference allele. This compares to 17,823 cases of significant TF–allele bias using a graph genome, with 52.8% favoring the reference allele (Supplemental Fig. S1A). We observed an average increase in total read mapping of 1.24% when using personalized graph genomes compared with the GRCh38 linear reference via Bowtie 2 (Supplemental Table S2; Supplemental Fig. S1B). We found that ASB events identified as significant using both alignment methods largely agreed on the direction of the effect (Supplemental Fig. S1C).
Overview of experimental design and allele-specific binding (ASB). (A) Workflow for detection of ASB. Whole-genome sequencing and ChIP-seq of 94 TFs were performed in postmortem brain samples from two donors. ChIP-seq reads were mapped to personalized graph genomes and tested for ASB across donors and tissues. Abbreviations denote the following: (DLPFC) dorsolateral prefrontal cortex, (FP) frontal pole, (OL) occipital lobe, (CB) cerebellum, (AnCg) anterior cingulate, (SCg) subgenual cingulate, (DMPFC) dorsomedial prefrontal cortex, (Amy) amygdala, and (HC) hippocampus. (B) Density plot showing the distribution of reference allele frequency at heterozygous variants for different P-value thresholds. (C) The number of total phased variants, heterozygous variants, significant variants (P < 0.05), and significant variants (P < 0.001) for both donors. “Shared” indicates variants that are shared and pass filters in both donors. (D) Within-donor reproducibility. The fraction of ASB events that preferred the same allele across brain regions at increasing P-value threshold. (E) Between-donor reproducibility. The fraction of ASB events that favored the same allele when comparing the same TF–allele interaction between donors.
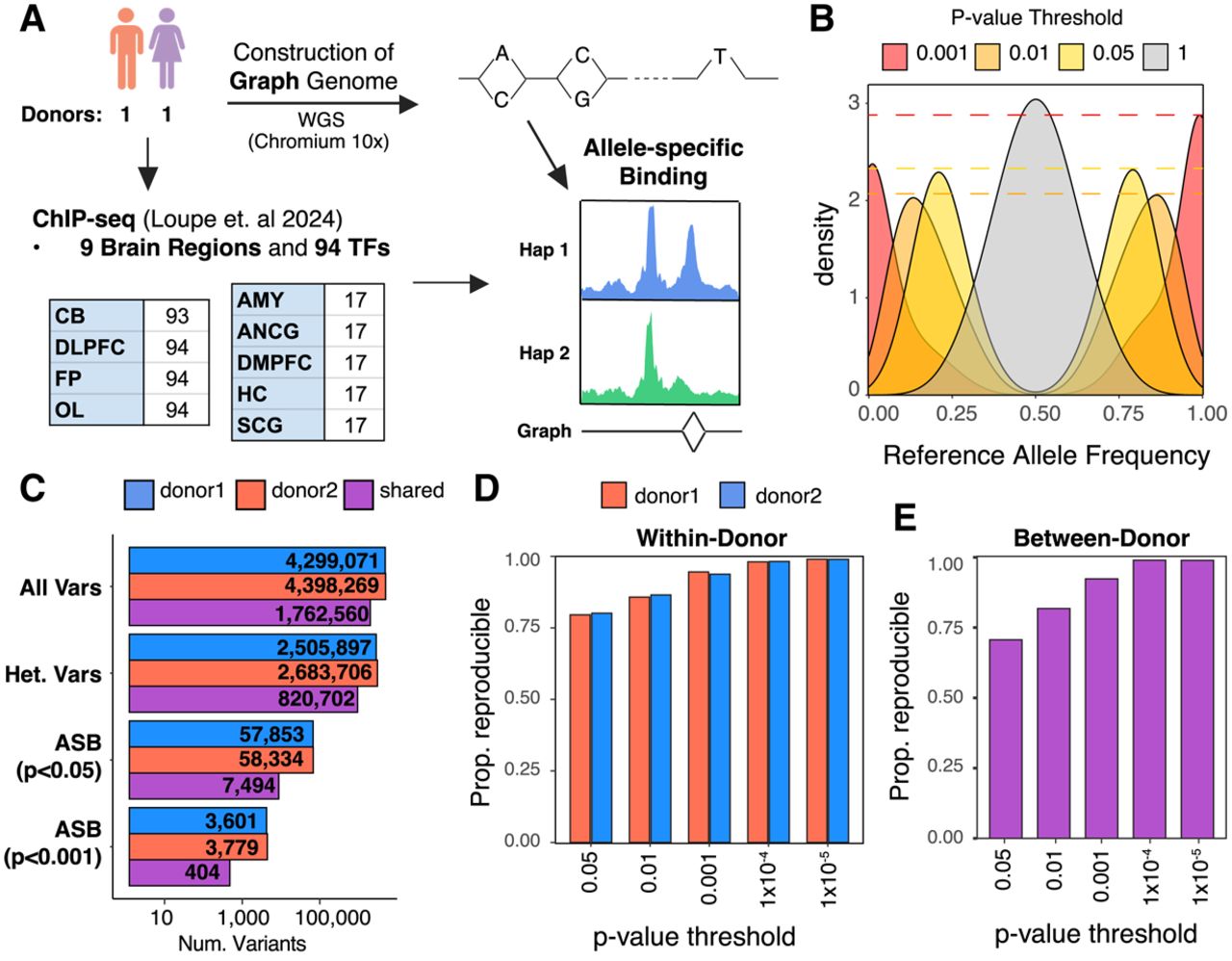
We then further corrected for allelic alignment bias using WASP (van de Geijn et al. 2015), which removes reads that fail to map to the same position after the heterozygous allele is flipped. This filter removed an average of 30,000 reads per ChIP-seq data set, primarily from genomic regions lacking prior evidence of regulatory function from ENCODE (Supplemental Fig. S1D,E). After these corrections, the average reference allele frequency at all heterozygous sites was 0.5, and 50.02% of nominally significant (P < 0.05) allele-specific variants favored the reference allele (Fig. 1B). At a more stringent threshold (P < 0.001), 57.5% of variants preferred the reference allele. However, this is likely reflective of true ASB, in which rare alternate alleles are more likely to exhibit decreased binding relative to the more prevalent allele (see below).
ASB significance and reproducibility analyses
We measured the significance of ASB using a beta-binomial test, applying it to ChIP-seq data from 94 TFs in up to nine anatomically defined regions of the brain across two donors, across a total of 920 ChIP-seq data sets (Supplemental Fig. S2A; Loupe et al. 2024). Among the more than 2 million heterozygous variants identified in each donor, 17% (Donor1: 505,637; Donor2: 432,723) have coverage of at least seven reads (the minimum needed to potentially achieve P < 0.05) for at least one TF in at least one tissue sample. Among those, 13% (Donor1: 57,853; Donor2: 58,334) exhibited nominally significant ASB for at least one TF (P < 0.05) (Fig. 1C). Among the variants that were heterozygous in both donors (207,086 variants), 30% of variants with at least seven reads (7494/25,215) exhibited ASB in both. We identified comparable numbers of ASB variants per TF in each donor with relatively low levels of dispersion across experiments (Supplemental Fig. S2A,B). The majority of ASB variants (70.5%) only impacted the binding of one TF (Supplemental Fig. S2C).
We next assessed the reproducibility of results between different brain regions within the same donor and from the same region between the two donors. True ASB should enrich for the same allele when measured in two independent experiments. Further, the rate of discordance in allelic effect directions between experiments allows one to measure the false-discovery rate (FDR), which is equal to twice the rate of discrepancies (because half of all false ASB discoveries will randomly show effect direction concordance). This empirical measure of reproducibility provides a robust measure of the effects of noise and does not require any assumptions about the underlying prevalence or effect size distribution of genuine ASB. We note that to the extent some observed ASB effect direction discrepancies may reflect genuine biological differences between experiments (e.g., two different donors), this would lead to these reproducibility estimates being conservative. First, for each P-value cutoff, we determined the proportion of significant TF–variant pairs that exhibited a preference for the same allele across different brain regions within each donor. We found that at a nominal P-value cutoff of 0.05, concordance is 79.8%, and at a P-value cutoff of 0.001, concordance increases to 94.2% (Fig. 1D; Supplemental Table S3); these correspond to empirical FDRs of 40% and 12%, respectively. We also measured between-donor reproducibility for shared heterozygous variants that were significant in at least one donor and had at least seven reads in both donors. For each P-value threshold, we determined what proportion of variants favored the same allele in ChIP-seq experiments from the same brain region in each of the two donors. Reproducibility between donors was lower than that within-donor but still substantial, being 70.6% reproducible at P < 0.05 and 92.3% at P < 0.001 (Fig. 1E; Supplemental Table S3). These values correspond to an empirical FDR of 59% and 15%, respectively. We also generated Benjamini–Hochberg (BH)-adjusted values based on the total numbers of heterozygous variants with at least seven reads in each experiment (Supplemental Table S4); the BH FDR estimates are higher and likely relatively conservative; and 2837 variants meet a BH-corrected P-value of 0.05. We confined further analyses of ASB to those variants with a nominal P-value < 0.001 (cross-donor FDR estimate of 15%, BH FDR 51%) unless noted otherwise.
Rare variation has a larger impact on the allele specificity of TF binding
We explored the genomic properties of the 6976 variants that impact TF binding at nominal P < 0.001 by utilizing candidate cis-Regulatory Elements (cCREs) from the ENCODE Consortium (The ENCODE Project Consortium et al. 2020), which annotates elements such as promoters and enhancers. Each heterozygous variant, TF ChIP-seq peak, and ASB variant were categorized into cCRE annotations (Fig. 2A; Supplemental Fig. S3A). As expected, we found that the majority of ChIP-seq peaks lie within cCRE regions, whereas the majority of heterozygous variation occurs outside of cCRE regions. Most cases (86.3%) of ASB fall within or near cCREs. Moreover, 78.8% of ASB variants were found within a peak called for that TF, and 87.5% within a peak for any TF. Relative to global peak location distributions, ASB exhibited a 1.9-fold enrichment for promoter-like signatures (PLSs; P-value < 2.2 × 10−16, chi-squared test). Additionally, a substantial number of ASB variants were identified in distal enhancer-like signatures (dELSs) and proximal enhancer-like signatures (pELSs). However, we note that these enrichments reflect, at least in part, our ability to detect ASB given the increased average read depth of ChIP-seq at promoters relative to enhancers (P-value < 2.2 × 10−16, t-test) (Supplemental Fig. S3B).
Genetic and genomic properties of ASB variants. (A) Stacked bar plots showing the fraction of regions which overlap a particular cCRE annotation for all heterozygous variants (top), ASB variants (middle), and all TF peaks (bottom). Red indicates promoter-like signal (PLS); orange, proximal enhancer-like signal (pELS); yellow, distal enhancer-like signal (dELS); blue, CTCF-only; pink, DNase-H3K4me3; and gray, non-cCRE regions. (B) Bar plot showing the proportion of ASB variants (P < 0.001) favoring the ancestral and derived allele and the degree of the allele preference [log2(Anc. count + 1/Der. count + 1)] for varying ranges of AAF. (C) For varying ranges of AAF (x-axis), we show the fraction of ASB variants found in each P-value range (y-axis). (D) The log2 fold enrichment (FE) for variants with a GERP-score > 4. The background for each TF is the nonsignificant heterozygous variants with a read depth greater than 11 for the corresponding TF. Only TFs with a chi-squared P-value < 0.01 are shown. (E) For each TF, the proportion of ASB variants that significantly alter a JASPAR motif (motifbreakR P-value < 0.0001) versus the maximum motif score for the PFM. Dashed line indicates the best fit line. (F) The proportion of ASB events that agree with the direction of the motif change in HOT sites versus non-HOT sites.
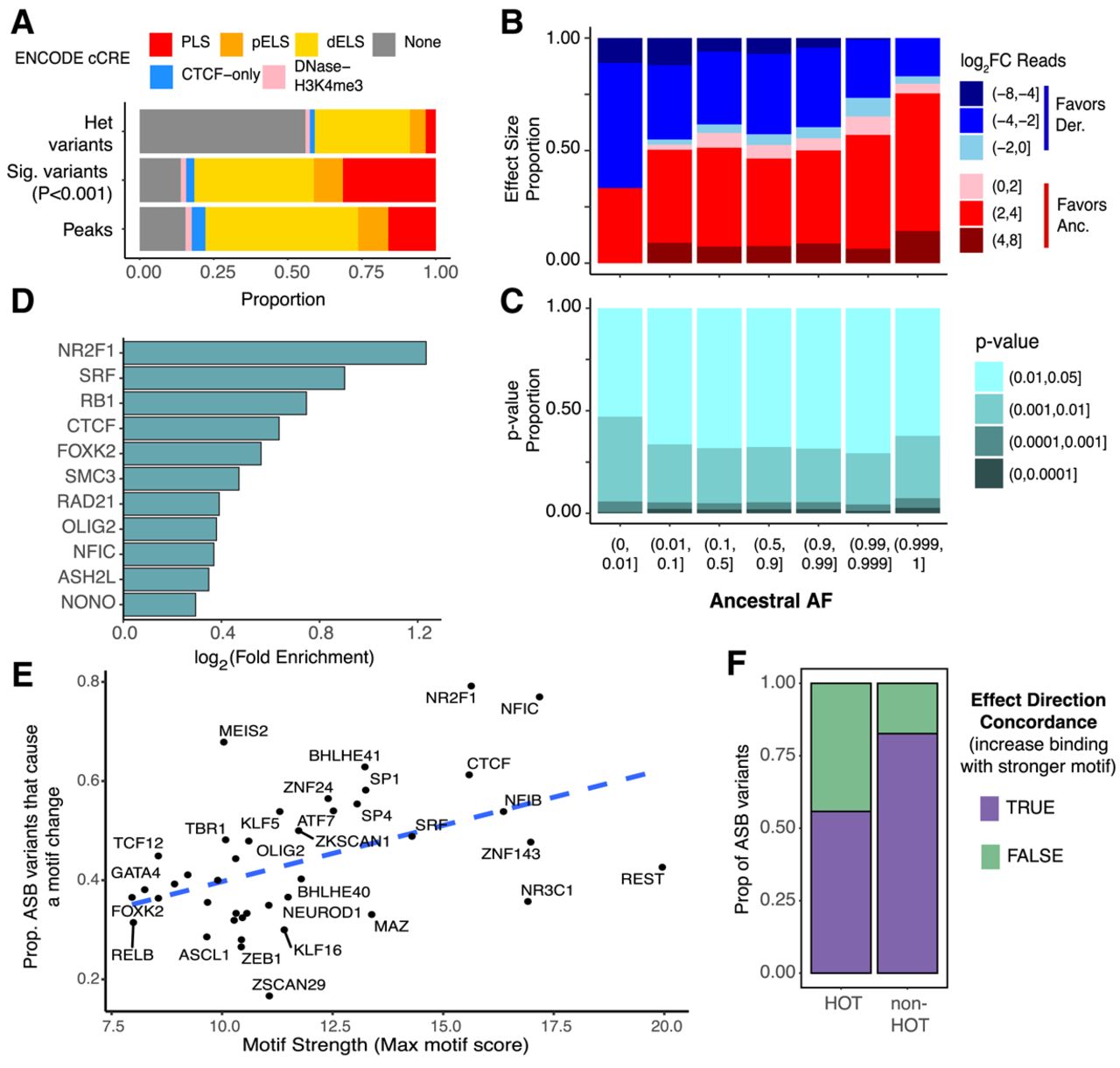
ASB variants are found in known regulatory regions, indicating potential functional implications for cellular and developmental phenotypes. Therefore, we investigated the prevalence of these variants in the population. We hypothesized that any derived allele that deleteriously altered the expression of a gene would undergo natural selection during human evolution. Using gnomAD v3 (Chen et al. 2024) to identify current allele frequencies in the human population and Ensembl (Cunningham et al. 2022) to identify ancestral alleles, we categorized ASB variants by ancestral allele frequency (AAF) and determined the proportion within each bin. We then measured the degree of specificity for the allele by calculating the log2 fold change (FC) in read counts for the ancestral and derived alleles. Variants with an AAF > 0.999 (DAF < 0.001) exhibited a pronounced preference for the ancestral allele, with <20% of variants favoring the derived allele. Notably, we observed an increase in the specificity for the derived allele when the ancestral allele was rare in the population (AAF < 0.01). (Fig. 2B). Conversely, among common variants with a AAF between 0.1 and 0.9, there is minimal bias toward either allele (ancestral: 54% vs. derived: 46%). Similarly, the major allele (gnomAD global frequency) was more strongly preferred, especially among very rare minor alleles (MAF < 1 × 10−5) (Supplemental Fig. S3C). Because false heterozygous calls will exhibit artifactual ASB, we also performed this analysis excluding all monoallelic ASB variants (i.e., 100% of ChIP-seq reads include only one allele) but found that the same trends in ancestral allele preference were observed (Supplemental Fig. S3D). We also looked at the distribution of P-values in the same AAF bins (Fig. 2C). Our analysis revealed that rare variation in the population (AAF ≤ 0.01) tended to be more significant. However, the distribution of P-values did not differ greatly between 0.01 < AAF ≤ 1. This suggests that common alleles are equally as likely to increase or decrease TF binding, whereas rare alleles are more inclined to disrupt TF–DNA association while consequently having a more pronounced impact on binding.
Observing these trends in allele frequency, we then investigated whether there was enrichment or depletion of ASB variants at sites that are conserved across mammalian species. To assess this, we used the genomic evolutionary rate profiling (GERP) (Davydov et al. 2010) metric and identified variants at genomic sites with GERP scores > 4, a commonly used cutoff for selective constraint (Schubert et al. 2014; Marsden et al. 2016). For each TF, we identified all variants with at least 11 reads mapped (the minimum number of reads needed to potentially achieve a beta-binomial significance of 0.001) and determined the proportion of variants with GERP scores greater than and less than four for both significant and nonsignificant ASB variants. (Supplemental Fig. S3E). Our analysis revealed that ASB variants for most TFs are not significantly enriched or depleted for conserved positions (chi-squared test). However, 11 TFs, including NR2F1 and CTCF, demonstrated significant enrichment for ASB variants at conserved sites (Fig. 2D). This shows that certain TFs have a higher association between mammalian conservation and ASB. Although this may reflect the effects of purifying selection, other confounders associated with sequence conservation, such as mutability and GC content, may also be relevant.
Relationships between motifs and ASB
To assess potential mechanisms driving ASB, we utilized motifbreakR and JASPAR 2022 human motifs to identify motifs affected by ASB variants (Supplemental Table S5; Coetzee et al. 2015; Castro-Mondragon et al. 2022). For TFs with a motif (43/94), we determined the proportion of the ASB variants that resulted in a change for the corresponding motif. This proportion varied across TFs, ranging from 26%–79% (Fig. 2E). This variability in part reflects the motif length and specificity of each TF. For example, NFIC, which has a 17 bp consensus motif containing many informative bases, had motif alterations at 77% of the ASB variants for that factor. In contrast, ZEB1, which has a 6 bp motif, had a motif altered in only 26% of the ASB variants. We then estimated a given motif's strength by taking the maximum possible motifbreakR score for the position frequency matrix (PFM). Motif strength was significantly correlated with the proportion of ASB variants for that TF that altered a motif (Pearson's correlation, r = 0.48, P = 0.0017).
Additionally, we examined the agreement in effect direction for motif alteration and binding preference. Across ASB sites for all TFs with motifs, the allele with the stronger motifbreakR score displayed preferred binding compared to the other allele (chi-squared test, OR = 4.40, P < 2.2 × 10−16). Further, 41/43 TFs (95%) with a motif showed nominal concordance of ASB and motif direction effects across their ASB sites. However, only five TFs individually showed a significant enrichment for effect direction concordance, including CTCF (log2FE = 6.47, P < 2.2 × 10−16), NFIC (log2FE = 5.38, P < 2.2 × 10−16), NFIB (log2FE = 5.03, P = 0.0005), NR2F1 (log2FE = 2.35, P = 0.0004), and SP1 (log2FE = 1.16, P = 0.002). The other TFs likely had too few ASB variants to observe a significant concordance (Supplemental Fig. S3F).
At least some discordance between motif effect and ASB may result from ASB sites in high occupancy target (HOT) sites (Wreczycka et al. 2019; Loupe et al. 2024). HOT sites have a higher abundance of TFs binding, are enriched for nonspecific TF binding, and are less dependent on the effects of any one motif. ASB variants within HOT sites are less likely than ASB variants outside HOT sites to specifically alter a motif (chi-squared test, OR = 0.76, P-value < 2.2 × 10−16). Furthermore, for the variants that do significantly alter a motif in a HOT site, they are less likely to have effect direction concordance than ASB variants outside of HOT sites (chi-squared, OR = 3.77, P-value < 2.2 × 10−16), supporting the hypothesis that motif changes at HOT sites are not driving changes in binding (Fig. 2F).
Variation of ASB across different brain regions
We assessed the overall correlation in ASB effects between different brain regions. For each pairwise comparison, we determined the correlation of both the nominal significance [−log10(P-value)] and the effect size (percentage of reads mapping to the reference allele) for significant ASB variants (Fig. 3A). All correlations between brain regions were significant for both metrics, ranging from 0.53 to 0.74 for significance and 0.49–0.72 for effect size (Spearman's correlation coefficient). The cerebellum showed significant but comparatively lower correlation with the other eight tissues. This is consistent with the fact that the cerebellum has a markedly different cellular makeup than other brain regions owing to the high proportion of granule cells (Andersen et al. 1992; Loupe et al. 2024).
Neural-specific effects of ASB. (A) Correlation of ASB effects across brain regions for variants with a P-value < 0.001 in at least one brain region. (Top) Correlation of significance [−log10(P-value)]. (Bottom) Correlation of effect size (percentage of reads mapping to the reference allele [% REF]). (B) Dot plot showing PC2 and PC3 from a PCA of the −log10(P-values) for all variants with a P-value < 0.001 in at least one brain region. Color denotes the brain region. Shape denotes if the TF is CTCF/RAD21 or another TF in the data set. (C) Distribution of the percentage of peaks containing an ASB by TF. Dashed line indicates the average percentage. Only TFs with at least 15,000 peaks are shown. (D) Enrichment for ASB variants in cell type–specific snATAC-seq peaks. Neuron indicates that the peak was called in excitatory-only, inhibitory-only, or excitatory and inhibitory neurons. Shared peaks were identified in two or more cell types, excluding those shared between neuronal cell types. (E) Results from LDSC measuring heritability of ASB with GWAS traits grouped by disease type. Heatmap indicates coefficient Z-score from sLDSC. Feature-trait combinations with a Z-score significantly larger than zero (one-sided Z-test, P < 0.01) are indicated with a numeric value reporting the enrichment score. Abbreviations denote the following: (SCZ) schizophrenia, (BIP) bipolar disorder, (MMD) major depression disorder, (ASD) autism spectrum disorder, (ADHD) attention deficit hyperactivity disorder, (ALS) amyotrophic lateral sclerosis, (PD) Parkinson's disease, (AD) Alzheimer's disease, (SLE) systemic lupus erythematosus, (MS) multiple sclerosis, (RA) rheumatoid arthritis, (IBD) inflammatory bowel disease, (BMI) body mass index.
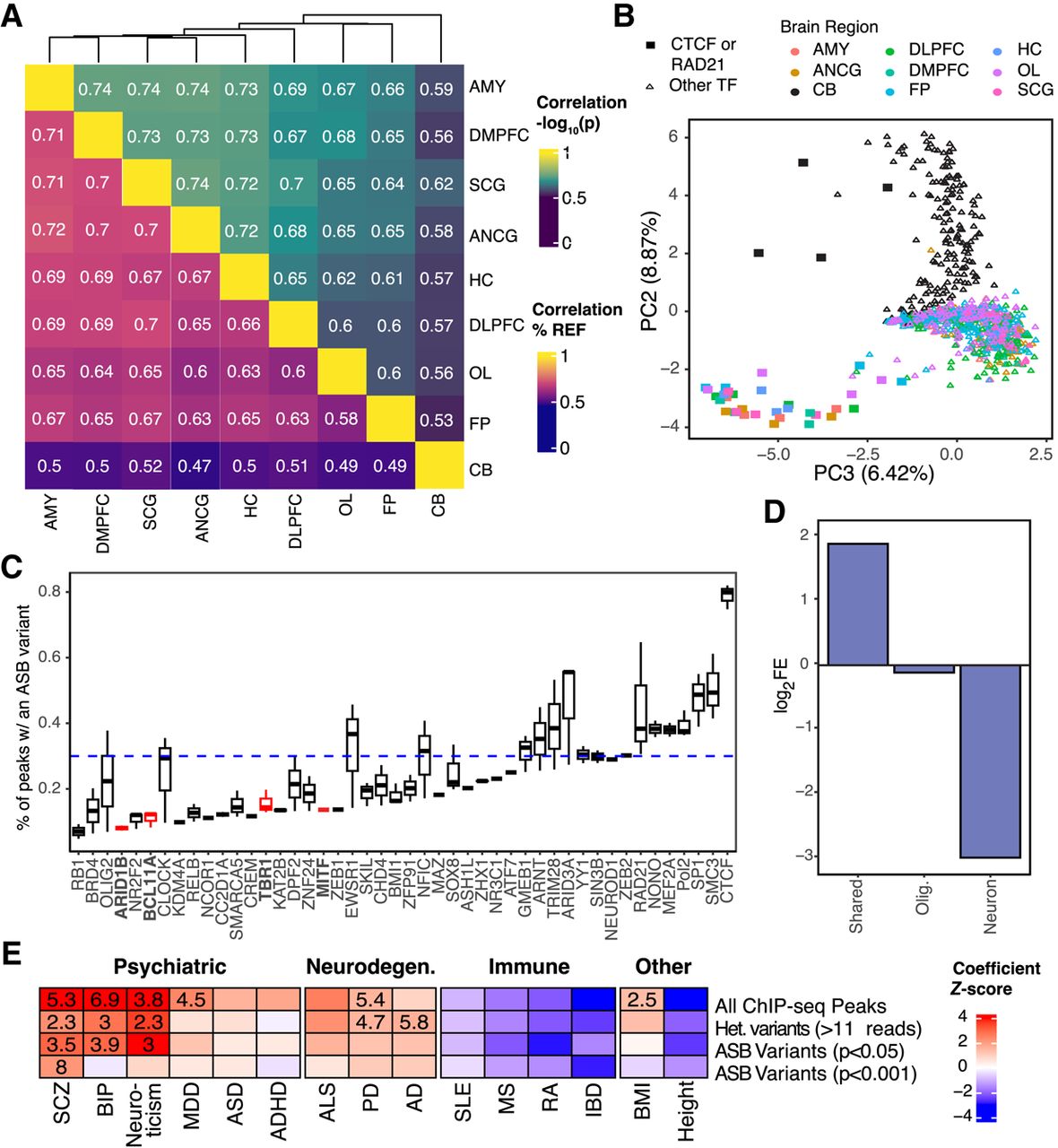
We further explored the difference between brain regions and TFs by performing a principal component analysis (PCA) on the −log10(P-values) for all variants that were nominally significant for at least one TF in at least one brain region. The first principal component (PC) is highly correlated with the number of peaks called for any given TF and did not provide much insight into the genome-wide differences in ASB between brain regions (Pearson's correlation, r = 0.82, P-value < 2.2 × 10−16) (Supplemental Fig. S4A). However, PC2 clearly separates cerebellum from the other brain regions and accounts for 8.87% of the variation in ASB across brain regions and TFs (Fig. 3B). More than 20% of ASB variants identified in CB were specific to the CB, whereas on average only 3% were specific to other individual brain regions (Supplemental Fig. S4B). Additionally, we can see that PC3 separates CTCF and RAD21 from all other TFs in this study. Although RAD21 does not have its own motif, it frequently colocalizes with CTCF (Hansen et al. 2017). As regulators of chromatin looping and domain boundaries, these TFs have a unique binding profile relative to other TFs and are influenced by a distinct set of variants (Loupe et al. 2024).
TF and cell type distribution of ASB
When we investigated which TFs were more likely to have occurrences of ASB, we found that the number of ASB variants for a TF was strongly correlated with the number of peaks called for that TF (Pearson's correlation, r = 0.83, P < 2.2 × 10−16) (Supplemental Fig. S4C). Across all TFs, the proportion of peaks with an ASB variant was on average 0.26%. However, there were certain TFs that had a notably lower incidence of ASB events, among which were key TFs involved in brain development, such as BCL11A (t-test, 0.12%, P-value = 0.0024), TBR1 (0.14%, P-value = 0.013), ARID1B (0.09%, P-value = 0.0006), and MITF (0.13%, P-value = 0.022) (Fig. 3C). To adjust for the fact that TFs may have variable rates of overlap with heterozygous variation (e.g., owing to distal vs. proximal CRE binding), we also estimated the rate of ASB as the proportion of peaks that overlap a heterozygous variant and observed the same trend for these TFs (Supplemental Fig. S4D).
To further assess the neuronal impact of ASB, we investigated the prevalence of ASB among different cell types. We assessed the enrichment for ASB variants within previously published cell type–specific and shared cell type snATAC-seq peaks generated from DLPFC, encompassing data from 15 donors, including the two donors analyzed in this study (Anderson et al. 2023). For this analysis, we specifically focused on variants that showed an ASB nominal P < 0.001 significance in DLPFC ChIP-seq experiments. We also only considered snATAC-seq cell type–specific peaks identified in neurons and oligodendrocytes to increase the likelihood that they would be detected in bulk tissue, given that these two cell types account for the majority of cells in DLPFC (Anderson et al. 2023). First, we observed an overall depletion of heterozygous variants in ChIP-seq peaks overlapping a neuron-specific snATAC-seq peak compared with other cell types (chi-squared, log2FE = −0.526, P < 2.2 × 10−16). Furthermore, neuron-specific peaks were significantly depleted for variants that caused ASB (log2FE = −2.36, P-value < 1.3 × 10−14). This cell type–specific depletion was not observed in oligodendrocytes for either heterozygous variants (log2FE = 0.12, P-value = 0.09) or ASB variants (log2FE = 0.18, P-value = 0.53) (Fig. 3D). Oligodendrocytes are at roughly similar, if not somewhat lower, abundances in DLPFC compared with neurons (Anderson et al. 2023). Furthermore, there is not a significant difference in read depth at neuron-specific and oligodendrocyte-specific ASB variants (t-test, P = 0.052) (Supplemental Fig. S4E), suggesting that lower read-depth at cell type–specific binding sites does not explain the depletion of ASB in neuron-specific peaks. Conversely, snATAC-seq peaks that were identified across multiple cell types were enriched for ASB variants (log2FE = 1.39, P-value < 9.0 × 10−10). This effect could not have been predicted by sequence conservation alone as we see no significant difference in GERP scores between nominally P < 0.001 significant and nonsignificant ASB variants in neuron-specific peaks (t-test, P-value = 0.32) or between cell types (P-value = 0.61).
ASB in genome-wide association studies
We performed stratified linkage disequilibrium score regression (sLDSC) (Finucane et al. 2015) to identify enrichments for variants detected in genome-wide association studies (GWAS) of neurodegenerative (Nalls et al. 2019; van Rheenen et al. 2021; Bellenguez et al. 2022) and psychiatric disease (Wray et al. 2018; Demontis et al. 2019; Grove et al. 2019; Mullins et al. 2021; Trubetskoy et al. 2022). We looked at the enrichment across all ChIP-seq peaks, heterozygous variants with binding (more than 11 ChIP-seq reads), ASB variants at P < 0.05, and ASB variants at P < 0.001. Notably, heterozygous variants with binding and ASB variants at P < 0.05 both exhibited significant enrichments for schizophrenia, bipolar disorder, and neuroticism, consistent with ChIP-seq on brain tissue enriching for regulatory elements relevant to these traits (Loupe et al. 2024). However, ASB variants showed a higher degree of enrichment for all three diseases (Fig. 3E; Supplemental Tables S6, S7). At P < 0.001, ASB variants were only significantly enriched for schizophrenia. This is likely because of the reduced power at this threshold, given that only 6976 variants reach this level of significance for ASB, resulting in a limited number of variants overlapping with GWAS hits for each disease. Notably, enrichments for non-brain-related GWASs were neither significant nor reached a nominally high degree of enrichment for any group of variants.
Evidence supporting ASB from other studies
We next investigated if ASB variants had evidence for impacting TF binding in other cellular contexts. We overlapped previously identified ASB variants from ADASTRA (Abramov et al. 2021), which is predominantly composed of ChIP-seq from cell lines. For TFs profiled in both studies, the average overlap in ASB variants for any given TF was 32%. However, the majority of overlap events involved different TFs, with CTCF (59%) and E2F1 (22%) being the only TFs that exhibited moderately specific overlap between the studies (Fig. 4A). For all other TFs, the majority of ASB variants that had previously been associated with ASB in other cell types impacted a different TF in the brain. Additionally, we overlapped our ASB variants with allele-specific open chromatin (ASoC) variants from induced pluripotent stem cell (iPSC)–derived neurons (Zhang et al. 2020) and found that 16% of the ASoC variants from that study that were also heterozygous in our donors also exhibited ASB. As has been previously reported, the overlap in epigenetic signatures between iPSC-derived neurons and postmortem brain tissue is limited due to iPSC reprogramming, making it difficult to estimate if this overlap is significant (Roessler et al. 2014; Penney et al. 2020). However, of those that overlapped, there was a highly significant enrichment for effect direction concordance (i.e., increased binding with increased accessibility; OR = 18.0, P-value < 2.2 × 10−16), suggesting replication across these distinct studies.
Functional characterization of ASB variants. (A) Proportion of ASB variants that overlap an ASB variant in ADASTRA for TFs profiled in both studies. (Light teal) Overlap occurs between the same TF between the studies; (dark teal) overlap occurs between different TFs. (B) Enrichment for effect direction concordance (i.e., increase in binding corresponds with increase in regulatory activity) with SuRE-seq data in K562 and HepG2 cells. Error bars indicate the 95% CI. (C) The odds ratio of effect direction concordance for GTEx eQTLs found in the brain and overlapping a significant ASB variant for various ASB P-value thresholds. Error bars indicate the 95% CI. (D) For each TF, the proportion of ASB variants that overlap an eQTL versus the proportion of those variants that agree on effect direction. (E) Histogram showing the distribution of distances from the TSS of an allele-specific gene to the closest ASB variant.
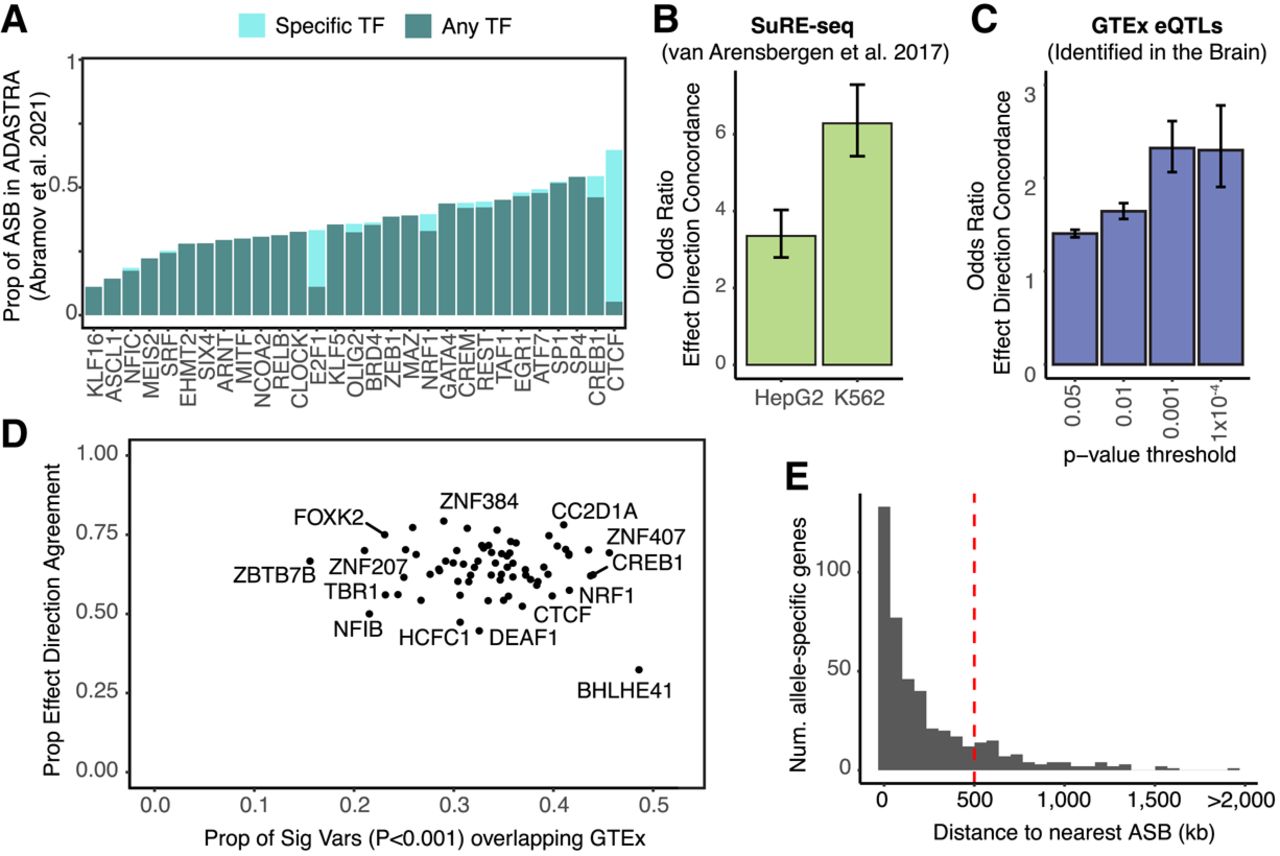
We next compared the sites of ASB we identified here with results in SuRE-seq data from K562 and HepG2 cells (van Arensbergen et al. 2017). Out of the 5.9 million variants observed by van Arensbergen et al., 48% were found to be heterozygous in at least one of our donors. Although this analysis is limited by the overlap in regulatory regions between the brain and K562/HepG2, which predominantly share TF binding sites at promoters (Loupe et al. 2024), 29% of ASB variants that were heterozygous in both studies also displayed a change in regulatory activity in either K562 or HepG2 cells. Furthermore, there was a high degree of concordance in the direction of the effect (i.e., increased binding with increased activity; K562: OR = 6.29, P-value < 2.2 × 10−16 HepG2: OR = 3.35, P < 2.2 × 10−16, chi-squared test) (Fig. 4B), underscoring that changes in TF binding correspondingly impact regulatory activity in reporter assays.
Given that TFs regulate the expression of RNA, we explored the association between ASB and allele-specific expression using the GTEx (GTEx Consortium 2020) database of expression quantitative trait loci (eQTLs) (Nica and Dermitzakis 2013). We found that 28.2% of all significant GTEx eQTL variants found in the brain were heterozygous in at least one of our donors (Donor1: 139,182; Donor2: 335,379). Of these variants, 35,635 variants had sufficient read coverage to determine ASB for at least one TF in this study, with 2670 variants displaying significant ASB at P < 0.001. For ASB variants, we assessed the agreement between the direction of gene expression change and the direction of the ASB at increasing ASB P-value thresholds. At all thresholds, we observed significant enrichment for concordance in effect direction (i.e., increased TF binding with increased gene expression, Fig. 4C). We also observed correlation between eQTL slope and ASB log2FC (Supplemental Fig. S5A), supporting the hypothesis that larger changes in binding lead to larger changes in expression. As we increased the ASB P-value threshold, we observed a higher enrichment for concordance of effect directions but a more than 10-fold decrease in the number of eQTLs overlapping an ASB variant (Supplemental Fig. S5B).
We next sought to identify individual TFs showing a stronger association with eQTLs. Notably, BHLHE41, CREB1, and ARID1B were among the TFs with the highest proportion of ASB variants overlapping an eQTL (Fig. 4D). For the majority of TFs, the proportion of ASB variants that agreed with the effect direction of the eQTLs exceeded 0.5. However, BHLHE41 was a clear outlier, with an effect direction agreement rate of only 32% (Fig. 4D). This is consistent with its known role as a transcriptional repressor (Pellegrino et al. 2014).
Comparison of ASB to RNA-seq
We next explored allele expression specificity by determining the number of reads preferring each allele for each tissue in RNA-seq generated for each of the two donors. Using a model of known protein-coding genes in the GRCh38 build (GENCODE v42), we determined which of these variants overlapped with annotated exons: 4.3% (Donor1: 109,762; Donor2: 108,445) of phased heterozygous variants occurred in known gene models, affecting 59% of genes. We detected allele-specific expression in 1.9% of gene bodies, consistent with estimates of the fraction of genes with allele-specific expression in earlier studies (Gimelbrant et al. 2007; Kravitz et al. 2023). Furthermore, when we overlaid this with ASB, we found that 68% (Donor1: 197/260; Donor2: 146/242) of genes with allele-specific expression occurred within 500 kb of an ASB variant, and the median distance from the TSS to the closest ASB variant was 110 kb (Fig. 4E).
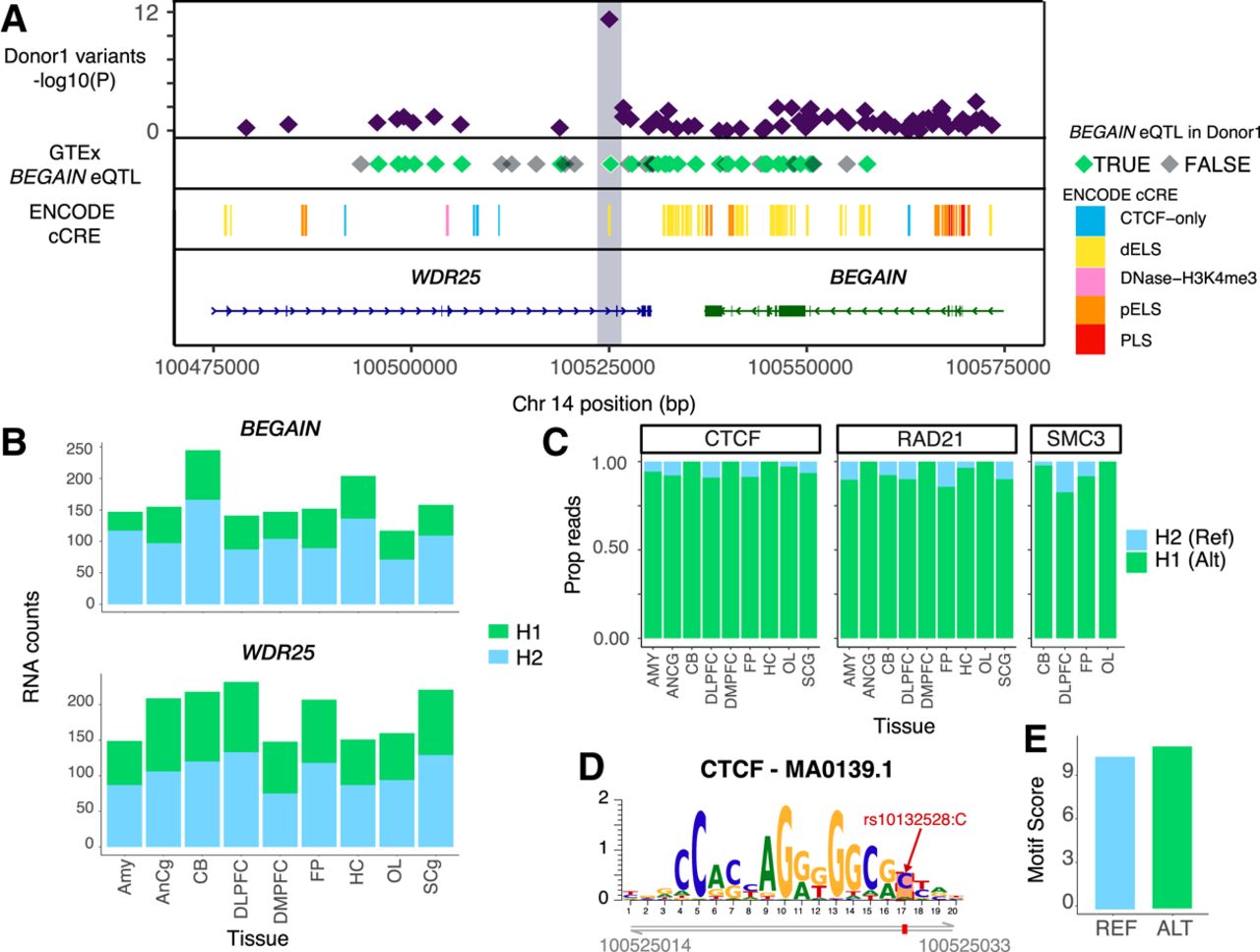
We highlight a case of ASB in Donor1, which coincides with a GTEx eQTL identified in the brain and demonstrates allele-specific expression in our data (Fig. 5A). The variant, Chr 14: 100525030:G/C, is located in the intron of WDR25 and exhibits ASB for cohesin proteins (CTCF, RAD21, SMC3). This variant is the only case of ASB in the locus across all TFs and overlaps an eQTL for BEGAIN (Fig. 5A). More than half of the significant eQTL variants identified for BEGAIN in GTEx are heterozygous in Donor1. All alleles that exhibited an increase in expression in GTEx were on the same haplotype in Donor1. In these data, we observe significant (P < 0.05) allele-specific expression of BEGAIN across brain regions, whereas WDR25 expression remains unaltered (Fig. 5B; Supplemental Table S8). CTCF, RAD21, and SMC3 all exhibit a strong preference for the alternate allele across all brain regions, with >90% of the reads aligning to it (Fig. 5C; Supplemental Table S4). When we look at the variant's effect on sequence recognition, we find that the alternate allele alters the CTCF-MA0139.1 motif and has a modest but significant increase in the motifbreakR motif score (allele effect size = 0.025, P-value = 4.5 × 10−6) (Fig. 5D). Consequently, this variant likely impacts the expression of BEGAIN through changes in 3D chromatin structure resulting from altered cohesin binding.
ASB analysis assists with fine-mapping eQTLs. (A) Genomic tracks for the 100 kb region surrounding rs10132528. (First) Maximum −log10(P-value) of all TFs for heterozygous variants in Donor1. (Second) GTEx eQTLs for BEGAIN colored by overlap with heterozygous variants in Donor1. (Third) ENCODE cCREs. (Fourth) Gene annotations from Ensembl v86. (B) Stacked bar plots showing the number of RNA reads supporting haplotype1 or haplotype2 for BEGAIN (top) and WDR25 (bottom). (C) The proportion of ChIP-seq reads for CTCF (left), RAD21 (middle), and SMC3 (right) supporting the reference and alternate allele for each brain region where the TF was profiled. (D) Position of the JASPAR CTCF motif on Chr 14 with the position of ASB variant indicated in red. (E) MotifbreakR score for the reference and alternate allele for rs10132528.
Discussion
Here, we present an analysis of ASB across 94 TFs, identifying thousands of variants that show ASB. We identified a threshold for reproducibility that provides confidence to our calls both within a single donor and between donors, controlling for a wide variety of biological and technical variables. By linking to allele-specific expression of nearby genes, we also relate variation that impacts TF binding directly to effects on gene regulation.
We found that rare variation (AAF > 99.9%/DAF < 0.1%) leads to stronger ASB (Fig. 3B), suggesting that there exists purifying selection against such variation in general. For common variants that do impact binding, neither the ancestral nor the derived alleles tend to be favored (Fig. 3C). In contrast, among rare variants (AAF > 99.9%/DAF < 0.1%), there is a strong bias in favor of the ancestral allele. This suggests that new mutations that lead to ASB more often disrupt, rather than increase, TF binding. The observation that ASB can sometimes prefer the derived allele is consistent with models of de novo motif formation and gene birth (Behrens and Vingron 2010; Carvunis et al. 2012; Ruiz-Orera et al. 2015; Schlötterer 2015; Papadopoulos et al. 2021; Iyengar and Bornberg-Bauer 2023).
We also found that many sites of ASB comprise multi-TF effects. Almost one-third (29.5%) of ASB variants influenced the binding of more than one TF (Supplemental Fig. S2C). Many of these are cohesin binding sites, as we found that ∼9.8% of ASB variants impacted two or more cohesin complex members. Consequently, we note that cohesin complex members had overall different profiles of ASB compared with other TFs, especially those that predominantly bind at highly occupied “HOT” sites (Fig. 2B). ASB variants affecting cohesin complex members also had a higher degree of overlap with eQTLs (Fig. 4B) and were significantly enriched for mammalian sequence conservation (Fig. 3E). This suggests that genetic variants that alter three-dimensional genome interactions are a major contributor to brain gene expression variation in the population, consistent with conclusions from previous studies of nonbrain tissues (Richer et al. 2023).
Although ASB is highly consistent across anatomically different brain regions, we did observe comparatively lower correlations to the cerebellum owing to its different cellular makeup compared with other brain regions (Fig. 3A). We also found that neuron-specific, unlike oligodendrocyte-specific, cCREs are depleted for ASB, whereas cCREs that are shared across brain cell types are enriched for ASB events. These results suggest that variation that leads to differences in binding, and consequently changes in neuronal gene expression, is selected against. However, this effect was not predicted by conservation alone as there were no significant differences in levels of mammalian sequence conservation for ASB variants found in cell type–specific cCREs and those found across cell types.
The enrichment for brain-related disease heritability in this data set highlights the value of studying ASB in a biologically relevant context. The increased enrichment of loci associated with psychiatric disorders in ASB variants relative to all heterozygous variants suggests that these regions contribute to neurological disease risk and are candidates for future study. The disparity in enrichment observed between schizophrenia and other psychiatric disorders in highly nominally significant ASB variants (P < 0.001) likely reflects the relatively lower number of variants detected at this level of ASB and supports the benefit of expanding this type of study to include more individuals so as to identify additional sites of ASB in brain tissue.
As has been previously observed (Abramov et al. 2021), we find that there is a general preference for a given TF to favor the allele with a stronger match to its motif (Supplemental Fig. S3C). Beyond demonstrating the general nature of this phenomenon, we identify those TFs that are most impacted by variation in their motif and those TFs that are more tolerant to novel motif creation. Motif alterations can then be combined with known eQTLs to facilitate fine-mapping and mechanistic hypothesis generation. For example, we highlight a case of a variant in an eQTL that displays ASB in our data set and specifically disrupts a motif for that TF (Fig. 5A–D). Although many variants exhibited allele-specific expression for the associated gene, only one variant also showed ASB. Furthermore, more than half of the eQTL variants associated with BEGAIN are heterozygous in Donor1, suggesting that although eQTLs are frequently confounded by variants in LD, ASB measurements can assist with the identification of causal variants within a locus. We identified 2670 GTEx eQTLs that also exhibited ASB for at least one TF, providing a rich resource for future eQTL fine-mapping efforts.
This study has several limitations. First, our ability to detect ASB is largely dependent on read depth. This affects our ability to identify cases of tissue-specific and cell type–specific ASB, as these enhancers specific to these contexts likely have lower read depth compared with those found in multiple cell types. Second, although the ChIP-seq data were produced and processed using ENCODE standards with as much uniformity as possible (e.g., large chromatin batches as inputs to the ChIPs) (Loupe et al. 2024), technical factors can influence comparisons between TFs, as is the case for all ChIP-seq analyses. Last, BH P-value corrections yielded larger FDR estimates (these are provided alongside the nominal P-values in Supplemental Table S4). We believe these values are overly conservative given the observed empirical reproducibility estimates, which are derived from comparing ASB across numerous pairs of independent experiments (Fig. 1D,E), but these more stringent values may be useful in applications in which higher specificity is valuable.
Overall, our study provides a resource of ASB variants that are experimentally shown to impact TF binding in brain tissue. These results further our understanding of how alteration in DNA sequence translates to changes in biological function, particularly in relation to analyses of gene regulation in the human brain and its effect on neurological disease.
Methods
Whole-genome sequencing and variant calling
We extracted high-molecular-weight DNA from ∼20 mg of cortex tissue from each donor using the MagAttract HMW DNA kit (Qiagen 67563). We prepared linked read libraries using the Chromium genome reagent kit v2 following the protocol provided by 10x Genomics. We processed sequence reads using the longranger software suite from 10x Genomics. We identified variants by aligning to a 10x Genomics-provided, longranger-enabled GRCh38 reference (version 2.1.0) using longranger wgs v2.2.2 (Marks et al. 2019). We called variants using GATK 3.8-1-0-gf15c1c3ef via the –vcmode GATK option in the longranger wgs workflow (Loupe et al. 2024). Using BCFtools query, heterozygous variants were filtered to those with an allele balance between 0.25 and 0.75 and with at least two reads for the ALT.
Genome construction
We constructed graph genomes using the vg toolkit version 1.20, available at GitHub (https://github.com/vgteam/vg) (Garrison et al. 2018). The “construct” command was used with the GRCh38 genome and all phased variants that passed quality metrics. We then pruned the graph using the “prune” command with default parameters. We produced the gbwt index using the “index” command with default parameters, and the gcsa index was created using the following parameters: -X 3 -Z 4000 -p -k 11.
RNA-seq
We performed RNA-seq for each of the nine brain regions as outlined by Loupe et al. (2024).
ChIP-seq experiments
We performed ChIP-seq experiments with 94 TFs in nine distinct brain regions. Full methods for production of ChIP-seq reads can be found in Loupe et al. (2024).
Peak calling
We called peaks according to the ENCODE Consortium's standard pipeline, using experiments from donors as replicates, as described by Loupe et al. (2024).
Read mapping
For traditional read mapping, we used Bowtie 2 (Langmead and Salzberg 2012) with default settings to map to the human GRCh38 genome.
For graph genome mapping, we used the vg map command with the arguments -A -K -M 3. The vg surject command was used to create SAM and BAM file formats for determining allele bias. The SAMtools package (Danecek et al. 2021) was used for sorting and filtering by quality. Picard was used for filtering duplicates.
Once reads were mapped and filtered, we used WASP (van de Geijn et al. 2015) to identify reads that showed a bias in mapping for either allele in the graph genome. The WASP “snp2h5” command was used to create an index of all heterozygous sites for each donor. Graph genome aligned GAMs were converted to BAMs in GRCh38 coordinates using “vg surject.” BAM files were intersected with heterozygous variants using the WASP “find_intersecting_snps.py” script, and reads overlapping more than two heterozygous variants were excluded. All other reads overlapping a heterozygous variant then had the allele flipped. For reads containing two variants, the alleles were flipped by haplotype. The flipped reads were then remapped to the graph genome with vg, as previously described, and then converted to GRCh38 coordinates. A custom script (intersect.R) was used to identify and remove reads that either no longer mapped or mapped to a different position when the allele was flipped. Reads with a vg MAPQ = 0 were also filtered.
Identification of allele bias
For a given ChIP-seq or RNA-seq data set, we counted the number of reads containing either allele for all heterozygous variants using the WASP script “bam2h5.” We then identified those heterozygous variants with at least six total reads (the minimum number of reads for a binomial test to be nominally significant at P < 0.05 if all reads map to a single haplotype). For each ChIP-seq data set, we estimated the overdispersion, rho, among variants with at least six reads by fitting a vector generalized linear model with a beta-binomial distribution using VGAM (Yee 2010). We then performed a beta-binomial test for each heterozygous variant using the corresponding estimate of rho from that ChIP-seq data set. We removed variants that showed significant allele specificity (P < 0.05) in the input control for any brain region. The ASB effect size was calculated as .
VEP annotations
We annotated VCF files for each donor using VEP. The config file is provided in the Supplemental_Code.zip file as vep108.ini. VEP engine and cache version 108 (McLaren et al. 2016) were used with a GRCh38 FASTA file. We used a merged transcript set of Ensembl (Cunningham et al. 2022) and RefSeq (O'Leary et al. 2016). Custom annotations were gnomAD (Chen et al. 2024) allele frequency using v3.1.1, Bravo TOPmed allele frequency freeze 8 (Taliun et al. 2021), GRCh38 GERP scores (as distributed with CADD v1.6), and CADD v1.6 scores (Rentzsch et al. 2019).
GTEx data and identification of allele-specific expression
We downloaded GTEx variants on June 30, 2023, from https://storage.googleapis.com/gtex_analysis_v8/single_tissue_qtl_data/GTEx_Analysis_v8_eQTL.tar.
For determining allele-specific expression in our data, we determined whether or not there was a phased heterozygous variant within the appropriate gene body in our data, as this was necessary for physically linking the ASB to allele-specific expression. In such cases, we determined the variant in the gene body that was on the same allele as each of the two haplotypes of the heterozygous variant in the GTEx data set. We counted the reads overlapping variants as described above and summed counts across exons and across brain regions. Significant allele-specific expression was calculated with a beta-binomial model. Effect size was calculated as .
Motif scoring
We predicted the effect of ASB variants on motif affinity using the R package motifbreakR and the catalog of JASPAR 2022 motifs for the corresponding TFs (Coetzee et al. 2015; Castro-Mondragon et al. 2022). MotifbreakR was run using the information content method, “ic,” and with equal background nucleotide frequencies (A = 0.25, C = 0.25, G = 0.25, T = 0.25). A P-value threshold of 0.0001 was used to filter significant motif changes.
Partitioned heritability analysis
To evaluate whether ASB variants are enriched with common genetic variants that have been associated with traits by GWAS, we performed stratified linkage disequilibrium score regression (sLDSC v1.0.1) (Finucane et al. 2015). sLDSC estimates the proportion of genome-wide SNP-based heritability that can be attributed to SNPs within a given genomic feature by a regression model that combines GWAS summary statistics with estimates of linkage disequilibrium from an ancestry-matched reference panel. GWAS summary statistics were downloaded for brain-related and other traits. The 200 bp region (−100, 100) around each ASB variant was tested individually. Rare variants were not excluded as these loci can still be associated with disease heritability. We used the full baseline model (baseline-LD model v2.2.) that included 97 categories capturing a broad set of genomic annotations. Links to GWAS summary statistics are available in Supplemental Table S7. Additional files needed for the sLDSC analysis were downloaded from https://alkesgroup.broadinstitute.org/LDSCORE/ following instructions at GitHub (https://github.com/bulik/ldsc/wiki).
Data analysis
We performed data analysis using R v4.1.0, as noted in appropriate scripts.
Downloaded data
We downloaded the V3 cCRE human data set from the ENCODE Portal. ADASTRA v5.1.3 data were downloaded from https://adastra.autosome.org/bill-cipher/downloads. ASoC data were downloaded from Supplemental Tables S4–S8.
Data access
All code used for these analyses is available via GitHub (https://github.com/aanderson54/BrainTF_ASB) and as Supplemental Code. All raw and processed sequencing data generated in this study are available through the PsychENCODE Consortium (https://doi.org/10.7303/syn4921369) under the accession number syn51942384.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This study was supported by National Institutes of Health grant 5R01MH110472 awarded to R.M.M. and G.M.C., the Memory and Mobility Fund from HudsonAlpha Institute for Biotechnology, and support from the Pritzker Neuropsychiatric Research Consortium. We thank the brain donors and their families without whom this research would not have been possible. We thank members of the Myers, Cochran, G. Cooper, and S. Cooper laboratories for many fruitful discussions and input.
Author contributions: Conceptualization was by R.M.M., G.M.C., and B.A.M. Investigation was by J.M.L. Data curation was by A.G.A., B.A.M., J.M.L., and J.M.J.L. Formal analysis was by A.G.A., B.A.M., J.M.L., I.R.-N., and S.A.F. Provision of brain tissue and resources was by W.E.B., B.G.B., P.M.C., A.S., S.J.W., and H.A. Writing was by A.G.A. and B.A.M. Supervision was by E.M.M. Funding acquisition was by R.M.M. and G.M.C.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278601.123.
References
- ↵Abramov S, Boytsov A, Bykova D, Penzar DD, Yevshin I, Kolmykov SK, Fridman MV, Favorov AV, Vorontsov IE, Baulin E, 2021. Landscape of allele-specific transcription factor binding in the human genome. Nat Commun 12: 2751. 10.1038/s41467-021-23007-0
- ↵Andersen BB, Korbo L, Pakkenberg B. 1992. A quantitative study of the human cerebellum with unbiased stereological techniques. J Comp Neurol 326: 549–560. 10.1002/cne.903260405
- ↵Anderson AG, Rogers BB, Loupe JM, Rodriguez-Nunez I, Roberts SC, White LM, Brazell JN, Bunney WE, Bunney BG, Watson SJ, 2023. Single nucleus multiomics identifies ZEB1 and MAFB as candidate regulators of Alzheimer's disease-specific cis-regulatory elements. Cell Genomics 3: 100263. 10.1016/j.xgen.2023.100263
- ↵Bailey TL, Johnson J, Grant CE, Noble WS. 2015. The MEME suite. Nucleic Acids Res 43: W39–W49. 10.1093/nar/gkv416
- ↵Behrens S, Vingron M. 2010. Studying the evolution of promoter sequences: a waiting time problem. J Comput Biol 17: 1591–1606. 10.1089/cmb.2010.0084
- ↵Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R, Grenier-Boley B, Andrade V, 2022. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet 54: 412–436. 10.1038/s41588-022-01024-z
- ↵Bonham LW, Sirkis DW, Yokoyama JS. 2019. The transcriptional landscape of microglial genes in aging and neurodegenerative disease. Front Immunol 10: 1170. 10.3389/fimmu.2019.01170
- ↵Carvunis A-R, Rolland T, Wapinski I, Calderwood MA, Yildirim MA, Simonis N, Charloteaux B, Hidalgo CA, Barbette J, Santhanam B, 2012. Proto-genes and de novo gene birth. Nature 487: 370–374. 10.1038/nature11184
- ↵Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Pérez N, 2022. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res 50: D165–D173. 10.1093/nar/gkab1113
- ↵Chen N-C, Solomon B, Mun T, Iyer S, Langmead B. 2021. Reference flow: reducing reference bias using multiple population genomes. Genome Biol 22: 8. 10.1186/s13059-020-02229-3
- ↵Chen S, Francioli LC, Goodrich JK, Collins RL, Kanai M, Wang Q, Alföldi J, Watts NA, Vittal C, Gauthier LD, 2024. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625: 92–100. 10.1038/s41586-023-06045-0
- ↵Clifton NE, Hannon E, Harwood JC, Di Florio A, Thomas KL, Holmans PA, Walters JTR, O'Donovan MC, Owen MJ, Pocklington AJ, 2019. Dynamic expression of genes associated with schizophrenia and bipolar disorder across development. Transl Psychiatry 9: 74. 10.1038/s41398-019-0405-x
- ↵Coetzee SG, Coetzee GA, Hazelett DJ. 2015. motifbreakR : an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 31: 3847–3849. 10.1093/bioinformatics/btv470
- ↵Cunningham F, Allen JE, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Austine-Orimoloye O, Azov AG, Barnes I, Bennett R, 2022. Ensembl 2022. Nucleic Acids Res 50: D988–D995. 10.1093/nar/gkab1049
- ↵Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, 2021. Twelve years of SAMtools and BCFtools. GigaScience 10: giab008. 10.1093/gigascience/giab008
- ↵Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. 2010. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6: e1001025. 10.1371/journal.pcbi.1001025
- ↵Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK. 2009. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25: 3207–3212. 10.1093/bioinformatics/btp579
- ↵Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, Baldursson G, Belliveau R, Bybjerg-Grauholm J, Bækvad-Hansen M, 2019. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet 51: 63–75. 10.1038/s41588-018-0269-7
- ↵Dror I, Golan T, Levy C, Rohs R, Mandel-Gutfreund Y. 2015. A widespread role of the motif environment in transcription factor binding across diverse protein families. Genome Res 25: 1268–1280. 10.1101/gr.184671.114
- ↵The ENCODE Project Consortium, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, 2020. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583: 699–710. 10.1038/s41586-020-2493-4
- ↵Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, Anttila V, Xu H, Zang C, Farh K, 2015. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47: 1228–1235. 10.1038/ng.3404
- ↵Garrison E, Sirén J, Novak AM, Hickey G, Eizenga JM, Dawson ET, Jones W, Garg S, Markello C, Lin MF, 2018. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat Biotechnol 36: 875–879. 10.1038/nbt.4227
- ↵Ghandi M, Mohammad-Noori M, Ghareghani N, Lee D, Garraway L, Beer MA. 2016. gkmSVM: an R package for gapped-kmer SVM. Bioinformatics 32: 2205–2207. 10.1093/bioinformatics/btw203
- ↵Gimelbrant A, Hutchinson JN, Thompson BR, Chess A. 2007. Widespread monoallelic expression on human autosomes. Science 318: 1136–1140. 10.1126/science.1148910
- ↵Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, 2019. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51: 431–444. 10.1038/s41588-019-0344-8
- ↵Groza C, Kwan T, Soranzo N, Pastinen T, Bourque G. 2020. Personalized and graph genomes reveal missing signal in epigenomic data. Genome Biol 21: 124. 10.1186/s13059-020-02038-8
- ↵GTEx Consortium. 2020. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369: 1318–1330. 10.1126/science.aaz1776
- ↵Hach F, Sarrafi I, Hormozdiari F, Alkan C, Eichler EE, Sahinalp SC. 2014. mrsFAST-Ultra: a compact, SNP-aware mapper for high performance sequencing applications. Nucleic Acids Res 42: W494–W500. 10.1093/nar/gku370
- ↵Hansen AS, Pustova I, Cattoglio C, Tjian R, Darzacq X. 2017. CTCF and cohesin regulate chromatin loop stability with distinct dynamics. eLife 6: e25776. 10.7554/eLife.25776
- ↵Huang G, Osorio D, Guan J, Ji G, Cai JJ. 2020. Overdispersed gene expression in schizophrenia. NPJ Schizophr 6: 9. 10.1038/s41537-020-0097-5
- ↵Iyengar BR, Bornberg-Bauer E. 2023. Neutral models of de novo gene emergence suggest that gene evolution has a preferred trajectory. Mol Biol Evol 40: msad079. 10.1093/molbev/msad079
- ↵Kravitz SN, Ferris E, Love MI, Thomas A, Quinlan AR, Gregg C. 2023. Random allelic expression in the adult human body. Cell Rep 42: 111945. 10.1016/j.celrep.2022.111945
- ↵Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, Weirauch MT. 2018. The human transcription factors. Cell 172: 650–665. 10.1016/j.cell.2018.01.029
- ↵Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923
- ↵Loupe JM, Anderson AG, Rizzardi LF, Rodriguez-Nunez I, Moyers B, Trausch-Lowther K, Jain R, Bunney WE, Bunney BG, Cartagena P, 2024. Multiomic profiling of transcription factor binding and function in human brain. Nat Neurosci 27: 1387–1399. 10.1038/s41593-024-01658-8
- ↵Marks P, Garcia S, Barrio AM, Belhocine K, Bernate J, Bharadwaj R, Bjornson K, Catalanotti C, Delaney J, Fehr A, 2019. Resolving the full spectrum of human genome variation using linked-reads. Genome Res 29: 635–645. 10.1101/gr.234443.118
- ↵Marsden CD, Ortega-Del Vecchyo D, Brien O, Taylor DP, Ramirez JF, Vilà O, Marques-Bonet C, Schnabel T, Wayne RD, Lohmueller RK, 2016. Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc Natl Acad Sci 113: 152–157. 10.1073/pnas.1512501113
- ↵Martiniano R, Garrison E, Jones ER, Manica A, Durbin R. 2020. Removing reference bias and improving indel calling in ancient DNA data analysis by mapping to a sequence variation graph. Genome Biol 21: 250. 10.1186/s13059-020-02160-7
- ↵McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. 2016. The Ensembl variant effect predictor. Genome Biol 17: 122. 10.1186/s13059-016-0974-4
- ↵Mimmack ML, Ryan M, Baba H, Navarro-Ruiz J, Iritani S, Faull RLM, McKenna PJ, Jones PB, Arai H, Starkey M, 2002. Gene expression analysis in schizophrenia: reproducible up-regulation of several members of the apolipoprotein L family located in a high-susceptibility locus for schizophrenia on chromosome 22. Proc Natl Acad Sci 99: 4680–4685. 10.1073/pnas.032069099
- ↵Mullins N, Forstner AJ, O'Connell KS, Coombes B, Coleman JRI, Qiao Z, Als TD, Bigdeli TB, Børte S, Bryois J, 2021. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat Genet 53: 817–829. 10.1038/s41588-021-00857-4
- ↵Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, 2019. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol 18: 1091–1102. 10.1016/S1474-4422(19)30320-5
- ↵Nica AC, Dermitzakis ET. 2013. Expression quantitative trait loci: present and future. Philos Trans R Soc Lond B Biol Sci 368: 20120362. 10.1098/rstb.2012.0362
- ↵O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44: D733–D745. 10.1093/nar/gkv1189
- ↵Ouyang N, Boyle AP. 2022. Quantitative assessment of association between noncoding variants and transcription factor binding. bioRxiv 10.1101/2022.11.22.517559
- ↵Papadopoulos C, Callebaut I, Gelly J-C, Hatin I, Namy O, Renard M, Lespinet O, Lopes A. 2021. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Res 31: 2303–2315. 10.1101/gr.275638.121
- ↵Paten B, Novak AM, Eizenga JM, Garrison E. 2017. Genome graphs and the evolution of genome inference. Genome Res 27: 665–676. 10.1101/gr.214155.116
- ↵Pellegrino R, Kavakli IH, Goel N, Cardinale CJ, Dinges DF, Kuna ST, Maislin G, Van Dongen HPA, Tufik S, Hogenesch JB, 2014. A novel BHLHE41 variant is associated with short sleep and resistance to sleep deprivation in humans. Sleep 37: 1327–1336. 10.5665/sleep.3924
- ↵Penney J, Ralvenius WT, Tsai L-H. 2020. Modeling Alzheimer's disease with iPSC-derived brain cells. Mol Psychiatry 25: 148–167. 10.1038/s41380-019-0468-3
- ↵Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. 2019. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47: D886–D894. 10.1093/nar/gky1016
- ↵Richer S, Tian Y, Schoenfelder S, Hurst L, Murrell A, Pisignano G. 2023. Widespread allele-specific topological domains in the human genome are not confined to imprinted gene clusters. Genome Biol 24: 40. 10.1186/s13059-023-02876-2
- ↵Roessler R, Smallwood SA, Veenvliet JV, Pechlivanoglou P, Peng S-P, Chakrabarty K, Groot-Koerkamp MJA, Pasterkamp RJ, Wesseling E, Kelsey G, 2014. Detailed analysis of the genetic and epigenetic signatures of iPSC-derived mesodiencephalic dopaminergic neurons. Stem Cell Rep 2: 520–533. 10.1016/j.stemcr.2014.03.001
- ↵Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, Leng J, Bjornson R, Kong Y, Kitabayashi N, 2011. AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol Syst Biol 7: 522. 10.1038/msb.2011.54
- ↵Ruiz-Orera J, Hernandez-Rodriguez J, Chiva C, Sabidó E, Kondova I, Bontrop R, Marqués-Bonet T, Albà MM. 2015. Origins of de novo genes in human and chimpanzee. PLoS Genet 11: e1005721. 10.1371/journal.pgen.1005721
- ↵Schlötterer C. 2015. Genes from scratch: the evolutionary fate of de novo genes. Trends Genet 31: 215–219. 10.1016/j.tig.2015.02.007
- ↵Schubert M, Jónsson H, Chang D, Der Sarkissian C, Ermini L, Ginolhac A, Albrechtsen A, Dupanloup I, Foucal A, Petersen B, 2014. Prehistoric genomes reveal the genetic foundation and cost of horse domestication. Proc Natl Acad Sci 111: E5661–E5669. 10.1073/pnas.1416991111
- ↵Smith RP, Taher L, Patwardhan RP, Kim MJ, Inoue F, Shendure J, Ovcharenko I, Ahituv N. 2013. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat Genet 45: 1021–1028. 10.1038/ng.2713
- ↵Stevenson KR, Coolon JD, Wittkopp PJ. 2013. Sources of bias in measures of allele-specific expression derived from RNA-sequence data aligned to a single reference genome. BMC Genomics 14: 536. 10.1186/1471-2164-14-536
- ↵Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, 2021. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature 590: 290–299. 10.1038/s41586-021-03205-y
- ↵Trubetskoy V, Pardiñas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, Bryois J, Chen C-Y, Dennison CA, Hall LS, 2022. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604: 502–508. 10.1038/s41586-022-04434-5
- ↵van Arensbergen J, FitzPatrick VD, de Haas M, Pagie L, Sluimer J, Bussemaker HJ, van Steensel B. 2017. Genome-wide mapping of autonomous promoter activity in human cells. Nat Biotechnol 35: 145–153. 10.1038/nbt.3754
- ↵van de Geijn B, McVicker G, Gilad Y, Pritchard JK. 2015. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat Methods 12: 1061–1063. 10.1038/nmeth.3582
- ↵van Rheenen W, van der Spek RAA, Bakker MK, van Vugt JJFA, Hop PJ, Zwamborn RAJ, de Klein N, Westra H-J, Bakker OB, Deelen P, 2021. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet 53: 1636–1648. 10.1038/s41588-021-00973-1
- ↵Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, Adams MJ, Agerbo E, Air TM, Andlauer TMF, 2018. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 50: 668–681. 10.1038/s41588-018-0090-3
- ↵Wreczycka K, Franke V, Uyar B, Wurmus R, Bulut S, Tursun B, Akalin A. 2019. HOT or not: examining the basis of high-occupancy target regions. Nucleic Acids Res 47: 5735–5745. 10.1093/nar/gkz460
- ↵Yee TW. 2010. The VGAM package for categorical data analysis. J Stat Softw 32: 1–34. 10.18637/jss.v032.i10
- ↵Zhang S, Zhang H, Zhou Y, Qiao M, Zhao S, Kozlova A, Shi J, Sanders AR, Wang G, Luo K, 2020. Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants. Science 369: 561–565. 10.1126/science.aay3983
- ↵Zhao G. 2023. Shared and disease-specific glial gene expression changes in neurodegenerative diseases. Nat Aging 3: 246–247. 10.1038/s43587-023-00378-1