Abstract
We report the construction and analysis of a new reference genome assembly for Rattus norvegicus, the laboratory rat, a widely used experimental animal model organism. The assembly has been adopted as the rat reference assembly by the Genome Reference Consortium and is named GRCr8. The assembly has employed 40× Pacific Biosciences (PacBio) HiFi sequencing coverage and scaffolding using optical mapping and Hi-C. We used genomic DNA from a male BN/NHsdMcwi (BN) rat of the same strain and from the same colony as the prior reference assembly, mRatBN7.2. The assembly is at chromosome level with 98.7% of the sequence assigned to chromosomes. All chromosomes have increased in size compared with the prior assembly and k-mer analysis indicates that the subject animal is fully inbred and that the genome is represented as a single haploid assembly. Notable increases are observed in Chromosomes 3, 11, and 12 in the prospective rDNA regions. In addition, Chr Y has increased threefold in size and is more consistent with the rat karyotype than previous assemblies. Several other chromosomes have grown by the incorporation of sizable discrete new blocks. These contain highly repetitive sequences and encode numerous previously unannotated genes. In addition, centromeric sequences are incorporated in most chromosomes. Genome annotation has been performed by NCBI RefSeq, which confirms improvement in assembly quality and adds more than 1100 new protein coding genes. PacBio Iso-Seq data have been acquired from multiple tissues of the subject animal and are released concurrently with the new assembly to aid further analyses.
The laboratory rat, Rattus norvegicus, provides an important model organism for genetic, genomic, and biological studies. Compared to the mouse, the adult rat has a 10 times greater body size that allows its use in physiological studies in which phenotype acquisition is challenging or less accurate in the smaller mouse. Further, the rat has a more complex set of behavioral capacities, including social behaviors, than the mouse. Some of these behaviors can be meaningfully associated with human analogs, and the rat has proven a useful model in studies of addiction and other behaviors (Solberg Woods and Palmer 2019; Bao et al. 2023; Zhou et al. 2023). As with the mouse, a number of inbred laboratory rat strains have been developed that provide the benefit of reduced complexity in genetic studies by generating animals with genomes that are effectively haploid. From these inbred lines, congenic and consomic lines have been bred to directly test genetic hypotheses (Russell 1985). In addition, recombinant inbred lines (Pravenec et al. 1989; Voigt et al. 2008) and heterogenous stocks have been produced from crosses between inbred strains (Hansen and Spuhler 1984), and panels of inbred rats have been assembled into hybrid diversity panels (Tabakoff et al. 2019) and are available as additional tools to pursue rat genetic studies.
From 2016 through 2020, there were about 40,000 PubMed entries per year in which rats were studied, indicating the scale of use of this model organism. The genetic underpinnings of these studies require a representation of the rat genome that is as complete and accurate as possible. An NIH-funded effort led by scientists at Baylor College of Medicine (BCM) in cooperation with the Rat Genome Sequencing Consortium and an additional private effort at Celera Genomics resulted in the first genome assemblies of the rat. The assembly completed by the Rat Genome Sequencing Consortium from a female of the inbred Brown Norway strain (BN/SsNHsdMcw) was adopted as the first rat reference assembly, RGSC 3.1, in 2004 (Gibbs et al. 2004). The assembly was represented in haploid ploidy. This was followed by several revisions and updates (RGSC 3.4, 2004 Rnor_4, 2010, Rnor_5, 2012 Rnor_6.0 2014). The Celera assembly (Rn_Celera) was released in 2006 and included sequence from both the Brown Norway and outbred Sprague Dawley strains. Assembly employed a hybrid approach combining whole-genome shotgun sequencing with bacterial artificial chromosome clone sequencing, resulting in highly fragmented assemblies. Rnor_6.0 sought to reduce fragmentation by introducing data obtained by newly developed long-read sequencing methods (Pacific Biosciences [PacBio] CLR).
The advancement of PacBio sequencing technology (reducing cost and increasing yield and read length) led to the production of a de novo BN/NHsdMcwi assembly created from PacBio continuous long reads (CLRs) that included additional methods (optical mapping, linked reads, and chromosome conformation capture) to facilitate scaffolding of assembled sequence contigs into longer-range structures (Howe et al. 2021b). This approach traded off read accuracy, a known limitation of the PacBio CLR method, against much better structural representation and resulted in the adoption of the mRatBN7.2 assembly by the Genome Reference Consortium (GRC) as the new rat reference assembly in 2020 (Howe et al. 2021b; de Jong et al. 2022). However, the mRatBN7.2 assembly approach considered that the Brown Norway strain reflected an outbred, diploid genome. This resulted in the application of methods to identify allelic variation and to separate reads representing presumed different haplotypes into an alternative pseudohaploid assembly. One consequence of applying this approach to a genome that is fully inbred and therefore effectively haploid is that what appears to be haplotypic variation may in fact represent sequences that are authentically duplicated in the genome but share sufficient similarity to be mistaken for alternative haplotypes.
The past 2 years have seen a remarkable period of progress in long-read sequencing technology. The advances incorporated into the mRatBN7.2 assembly as well as its limitations have been analyzed and reported (de Jong et al. 2022, 2024). These limitations of the mRatBN7.2 assembly motivated our current effort to restore the rat genome assembly to a haploid genome with contiguity, completeness, and accuracy that can be obtained by combining the more recent higher accuracy PacBio HiFi sequencing method with additional scaffolding and curation methods. Here we report the creation of a new rat reference genome assembly from a male Brown Norway of the same substrain used in prior assemblies (BN/NHsdMcwi). This assembly has been adopted as the new rat reference genome assembly by the GRC and named GRCr8.
Results
Assembly
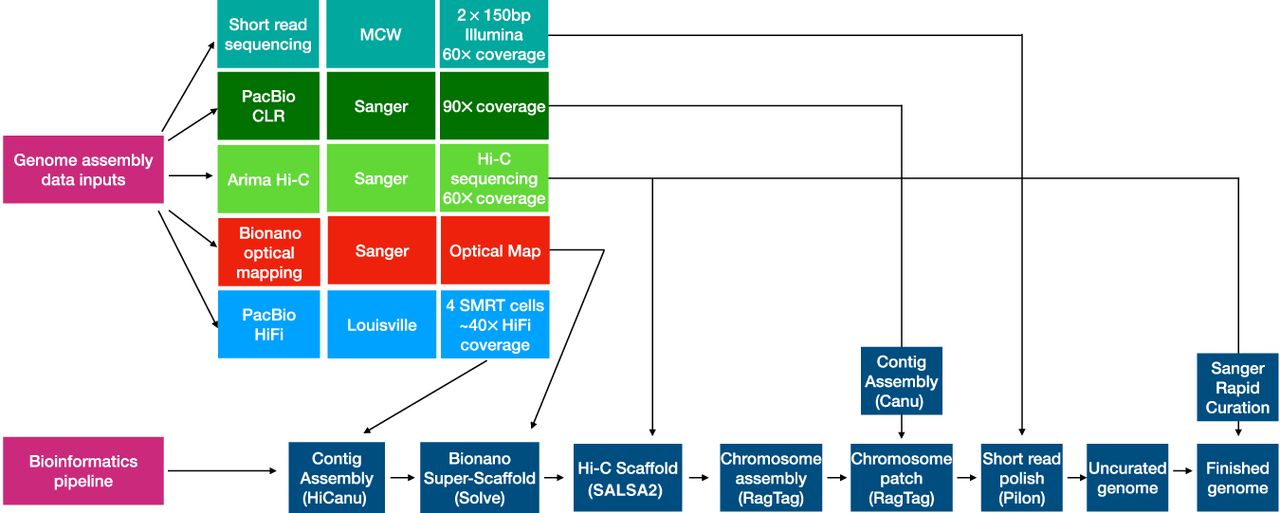
The data sources and sequential steps used to generate the genome assembly are outlined in Figure 1. These include HiFi sequencing, the use of existing optical mapping and chromosomal conformation data generated in the production of mRatBN7.2, reference-assisted assembly of subchromosomal scaffolds into single chromosomes (Alonge et al. 2022), assembly of existing CLR data into contigs for use in assembly gap filling, polishing of the assembly with Illumina short reads, and rapid curation of the assembly using Hi-C heat maps to visualize and correct misassemblies (Howe et al. 2021a).
Genome assembly pipeline and data inputs. Illumina short-read sequencing was generated by the Medical College of Wisconsin (MCW) and used for assembly polishing. PacBio CLR reads generated by the Wellcome Sanger Institute and originally used for creating the preceding reference assembly mRatBN7.2 were assembled with Canu, and the resulting contigs were used for assembly gap filling. Scaffolding was performed using Bionano Genomics optical reads and Arima Genomics Hi-C data generated at Sanger for the mRatBN7.2 assembly. PacBio HiFi reads were generated on a Sequel IIe instrument at the University of Louisville. These were assembled with HiCanu, and the resulting contigs were integrated with the Bionano optical map to create a hybrid assembly using Bionano Solve software at Texas A&M University. The Hi-C data were used to further scaffold the Bionano hybrid assembly using SALSA2 software. Reference-assisted chromosome assembly used RagTag software, and the existing mRatBN7.2 assembly served as a template to order and assemble scaffolds, into which the PacBio CLR contigs were incorporated for gap filling using the RagTag patch module. After polishing, the assembly was curated using the Sanger Institute's rapid curation pipeline, which uses Hi-C contact mapping to identify and resolve misassemblies.
Our assembly of the Brown Norway genome has resulted in chromosome-level assembly with a total extent (assembly assigned to chromosomes, unlocalized and unplaced scaffolds) of 2.8496 Gb. Of this, 98.7% was assigned to chromosomes, with a scaffold N50 of 137 Mb and a scaffold L50 of 8. The comparisons reported in the present paper are between the GRCr8 assembly and the mRatBN7.2 primary assembly haplotype. We examined the HiFi reads to identify a mitochondrial genome (ChrM) within the reads using MitoHiFi software (Uliano-Silva et al. 2023). This yielded a mitochondrial chromosome that was identical to that of mRatBN7.2 except that the new assembly contained a two base insertion immediately after position ChrM:5175 of the mRatBN7.2 assembly.
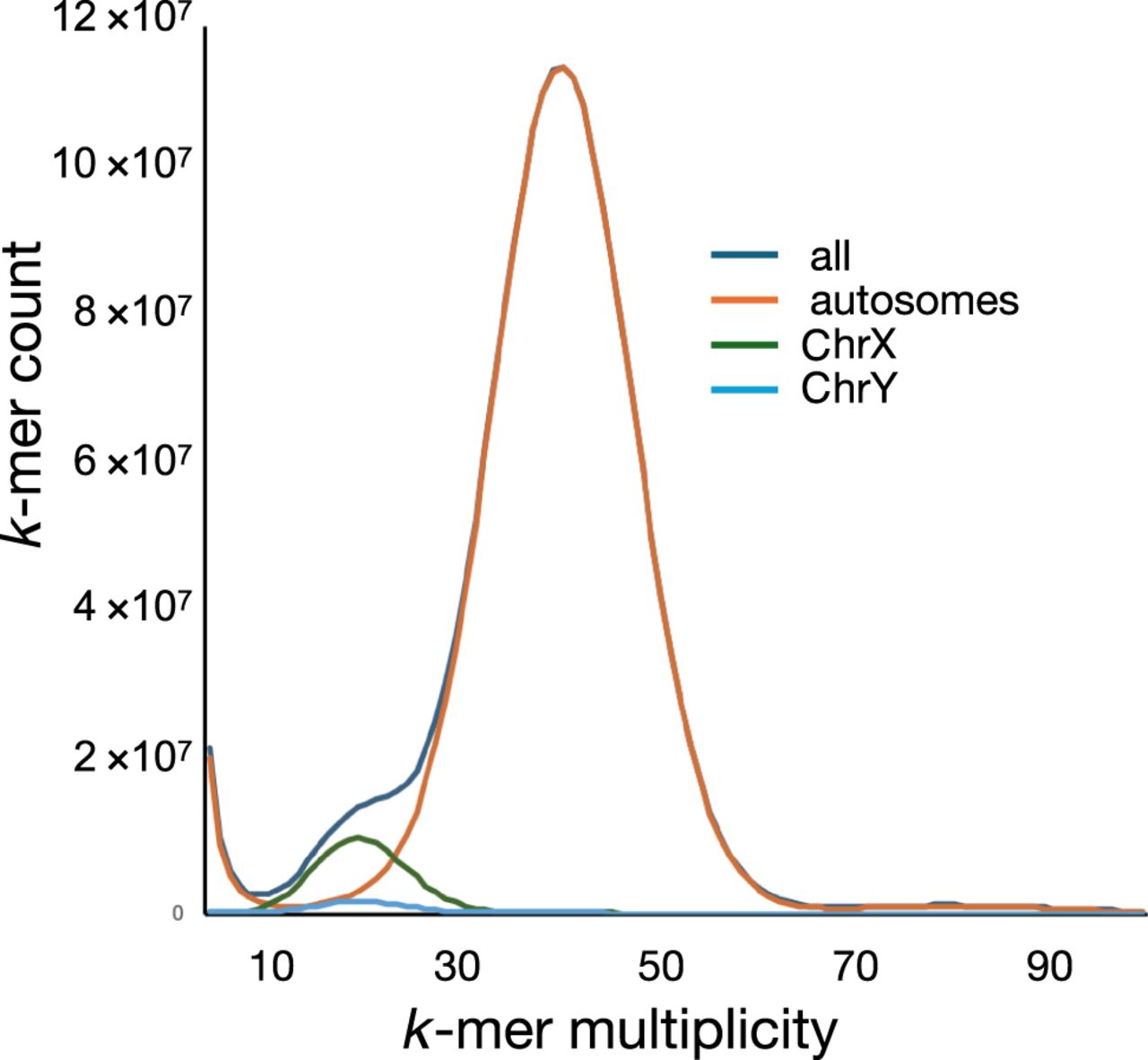
The assembly was analyzed by the NCBI Foreign Contamination Screen (FCS) tool and Genome Cross-species aligner (GX) to determine whether the assembly was contaminated with extrataxonic sequences (Astashyn et al. 2024). None were found. We applied Merqury analysis to characterize aspects of the assembly using k-mer analysis (Rhie et al. 2020). Figure 2 indicates the k-mer multiplicity of the assembly as determined by Merqury analysis. The initial k-mer multiplicity plot revealed two k-mer multiplicity peaks at 20 and 40. These peaks reflect k-mer counts associated with the sex chromosomes (the subject animal was male) and the autosomes, respectively. However, the presence of residual heterozygosity in this inbred strain might lead to autosomal k-mers in the 20-peak. To assess residual heterozygosity, we extracted all reads from the BN HiFi data set that mapped to the autosomes of the GRCr8 assembly. We then generated a k-mer multiplicity profile from these reads. Plotting the coverage depth against the k-mer frequency we saw a maximum at the expected 40× coverage. The presence of heterozygosity would result in k-mers with a coverage maximum of 20×. Neither a k-mer peak nor shoulder was observed in this region. We also examined k-mer spectra derived from BN HiFi reads mapped to Chr X and Chr Y, respectively, and demonstrate that the shoulder seen in the combined histogram is caused entirely by them (Supplemental Fig. S1). Merqury also identifies k-mers that are found only in the assembly and not in the reads, indicating assembly errors; none were observed. Finally, Merqury provides an estimate of assembly completeness by comparing the total number of k-mers in reads versus the total number in the assembly. Completeness was estimated at 100%. Assembly completeness was also assessed using conserved universal single copy orthologs (BUSCO) of the Glires (rodents and lagomorphs) clade (Waterhouse et al. 2018). The recently released software compleasm was used for this assessment (Huang and Li 2023). This employs protein-to-genome alignment for the assessment of completeness. Both mRatBN7.2 and the current assembly were estimated to be 99.7% complete as represented by the inclusion of highly conserved single-copy orthologs in the assembly.
We extracted all 21-mers from the HiFi reads to evaluate the k-mer distribution in the assembly. k-mers were aligned to the autosomes, extracted, and plotted to demonstrate multiplicity of k-mers. As anticipated, autosomally mapped k-mers show a single peak with a multiplicity similar to the sequencing read depth, indicative of the absence of heterozygosity. Similar mapping and plotting was done for the sex chromosomes, which both show a multiplicity indicative of their haploid state in the male subject genome. Supplemental Figure S2 shows each plot (autosomes, Chr Y, and Chr X) independently as well as the combined plot of all k-mers, allowing closer examination.
Identification of new components of the assembly
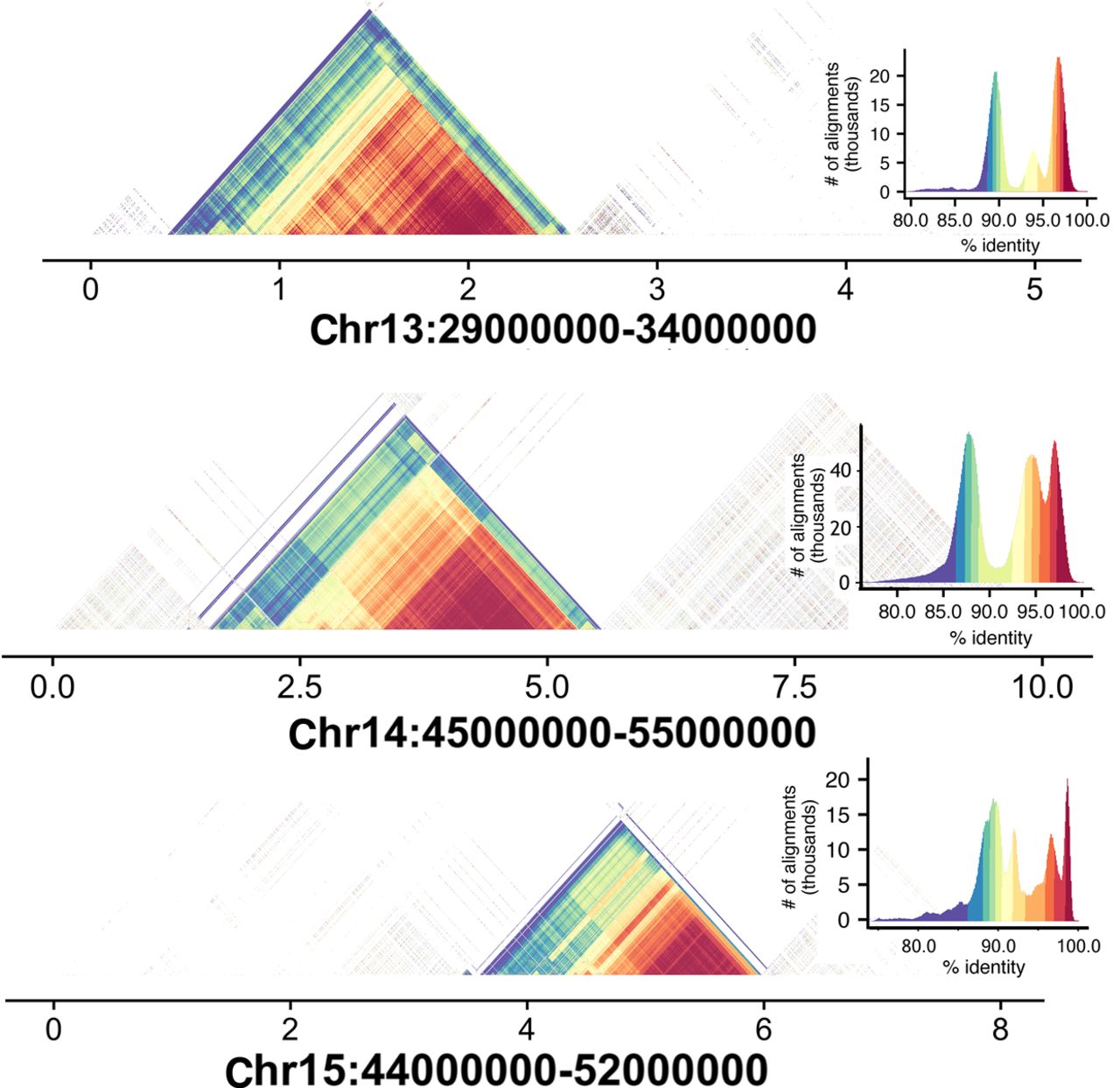
The introduction of HiFi sequencing to rat genome assembly indicated the possibility that regions of the genome with known low complexity and a high degree of repetitive sequences that were previously inaccessible might now be incorporated into the assembly. This was the case for a HiFi-based HiCanu assembly of the haploid human cell line CHM13, derived from a hydatidiform mole (Nurk et al. 2020). This assembly resolved nine of 23 expected centromeric regions. To assess the incorporation of centromeric regions in the rat assembly, we used StainedGlass software (Vollger et al. 2022) to reveal repetitive sequence in chromosomal regions expected to harbor centromeres, based on the published rat karyotype (Hamta et al. 2006). Unlike the mouse, which possesses only telomeric centromeres, distribution of rat centromeres are telocentric, acrocentric, and (sub)metacentric (see Table 1). Table 1 also indicates whether a distinct centromeric structure was detected by the StainedGlass analysis. In general, metacentric and submetacentric chromosomes had readily detectable centromeric repeat sequences that ranged in extent from 0.5 to 2 Mb (Fig. 3; Table 1). Centromere identification was more challenging in the acrocentric and telocentric chromosomes. Additional analysis of the centromeric and other noncentromeric repetitive regions added to this assembly is provided in Supplemental Figure S3. We also analyzed telomeric regions to determine whether the assembly had captured telomeric hexamer repeat sequences (Table 2). Telomeric hexamer repeats were indeed detected and found to be extensive in the metacentric chromosomes as well as in the distal terminals of acrocentric and telocentric chromosomes. Hexamer repeats were less often detected in the telomeres of telocentric chromosomes. Altogether this indicates that our assembly approach has been successful in incorporating a large amount of highly repetitive genome sequences that were not similarly captured in prior assemblies.
We used the StainedGlass software tool to assess self-to-self similarity in regions of the chromosome in which the rat karyotype indicates the likely location of centromeric satellite sequences. The darkest red colors indicate near 100% identity, and purple colors indicate identity up to 90%. For each analysis, the distribution of self-similarity across the region is indicated in the graph relating number of alignments observed with the specified similarity. For illustration, we show regions of three metacentric chromosomes in which clear self-similarity is present. For more details on other chromosomes, see Table 1.
Localization of repetitive centromeric structures in the assembly
| Chr | Centromere detected | Position (Mb) | Extent (Mb) |
|---|---|---|---|
| Chr 1 (SM) | N | ||
| Chr 2 (T) | Y | 2.5 | 0.5 |
| Chr 3 (A) | N | ||
| Chr 4 (T) | Y | 0.9 | 0.4 |
| Chr 5 (T) | Y | 2.3 | 4.4 |
| Chr 6 (T) | Y | 2.5 | 5 |
| Chr 7 (T) | Y | 0.2 | 0.4 |
| Chr 8 (T) | Y | 2.5 | 5 |
| Chr 9 (T) | N | ||
| Chr 10 (T) | Y | 0.3 | 0.4 |
| Chr 11 (A) | Y | 1.8 | 3.5 |
| Chr 12 (A) | N | ||
| Chr 13 (SM) | Y | 30.5 | 2 |
| Chr 14 (M) | Y | 48.5 | 4 |
| Chr 15 (M) | Y | 49 | 2.5 |
| Chr 16 (M) | Y | 42 | 1.6 |
| Chr 17 (M) | Y | 45 | 4 |
| Chr 18 (M) | Y | 38.6 | 1.8 |
| Chr 19 (M) | Y | 39 | 0.6 |
| Chr 20 (M) | Y | 26.2 | 0.5 |
| Chr X (T) | Y | 2.4 | 1.5 |
| Chr Y (T) | N |
[i] (A) Acrocentric, (M) metacentric, (SM) submetacentric, (T) telocentric, (Y) yes, and (N) no.
Telomeric sequences in the GRCr8 assembly compared with mRatBN7.2
| Proximal telomere size (kbp) | Distal telomere size (kbp) | ||||
|---|---|---|---|---|---|
| Telo/Acro/Metacentric | mRatBN7.2 | GRCr8 | mRatBN7.2 | GRCr8 | |
| Chr 1 | Metacentric | 0 | 9.2 | 2.8 | 13 |
| Chr 2 | Telocentric | 0 | 12 | 4.2 | 4.9 |
| Chr 3 | Acrocentric | 0 | 0 | 5.3 | 21.9 |
| Chr 4 | Telocentric | 0 | 7.2 | 4.6 | 12 |
| Chr 5 | Telocentric | 0 | 16.3 | 3.9 | 11.5 |
| Chr 6 | Telocentric | 0 | 0 | 3.7 | 23.3 |
| Chr 7 | Telocentric | 0 | 0 | 1.9 | 9.9 |
| Chr 8 | Telocentric | 0 | 0 | 3.5 | 8.3 |
| Chr 9 | Telocentric | 0 | 0 | 1.6 | 13.7 |
| Chr 10 | Telocentric | 0 | 0 | 2.2 | 6.7 |
| Chr 11 | Acrocentric | 0 | 0 | 2.8 | 10.6 |
| Chr 12 | Acrocentric | 0 | 0 | 0 | 13.9 |
| Chr 13 | Metacentric | 1.7 | 10 | 3.2 | 17.4 |
| Chr 14 | Metacentric | 3.2 | 18.2 | 5.9 | 8.8 |
| Chr 15 | Metacentric | 2.5 | 0 | 2.4 | 18.8 |
| Chr 16 | Metacentric | 3 | 9.9 | 2.1 | 14.4 |
| Chr 17 | Metacentric | 2.3 | 8.1 | 12.4 | 10.4 |
| Chr 18 | Metacentric | 1.6 | 15.9 | 6.5 | 22.4 |
| Chr 19 | Metacentric | 2.4 | 8.8 | 2.9 | 14.5 |
| Chr 20 | Metacentric | 2 | 7.3 | 1.4 | 5.1 |
| Chr X | Telocentric | 0 | 0 | 0 | 0 |
| Chr Y | Telocentric | 0 | 0 | 0 | 0 |
[i] Telocentric indicates that the centromere is at the proximal region of the chromosome. Acro/rDNA indicates an acrosomal centromere with rDNA's in the acrocentric arm. Metacentric indicates a meta- or submetacentric centromere. Telomere size (kbp) is estimated as the number of telomeric bases composed of hexamer repeat sequences.
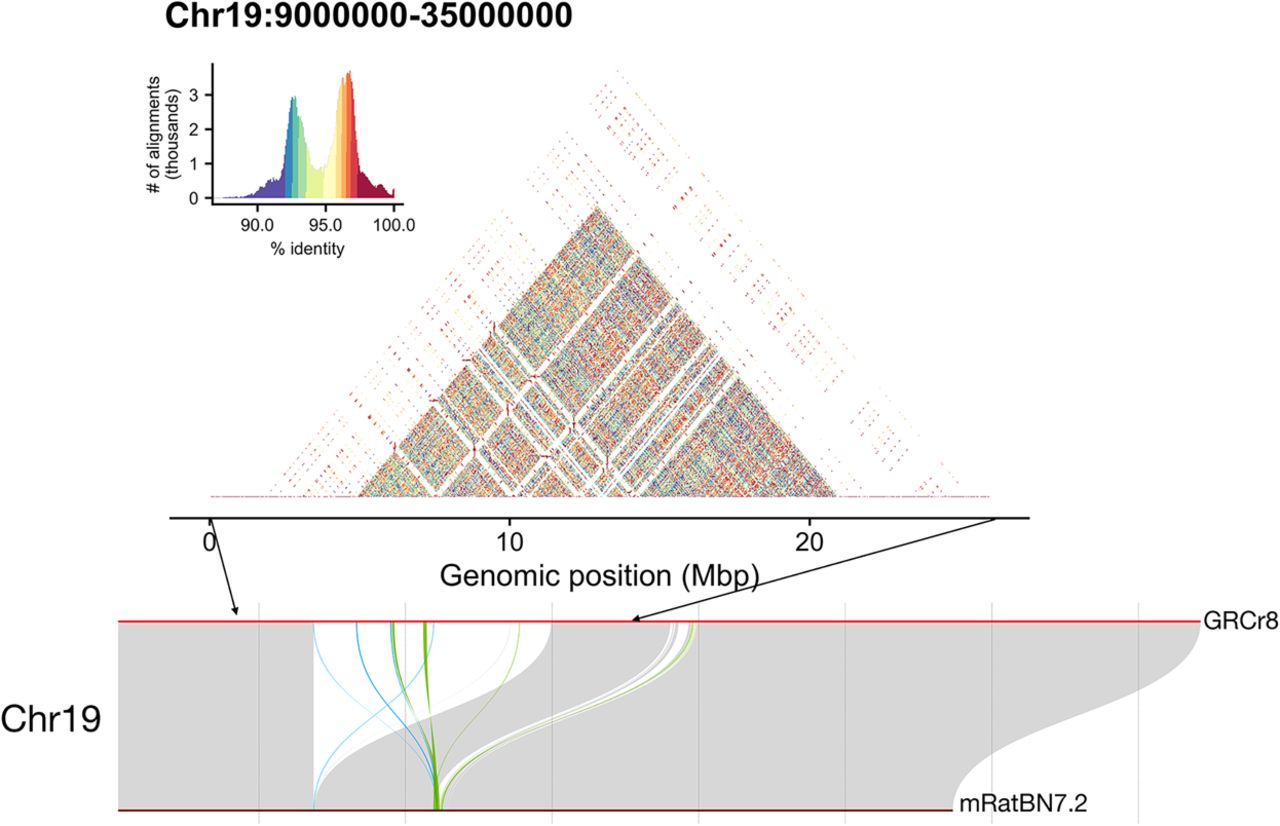
Table 3 lists the assembled chromosomes and compares their extent with those of the prior reference assembly mRatBN7.2. Notably all chromosomes have increased in size; however, the increases are not uniform in scale. Some chromosomes have increased more notably. Most obvious among these is Chromosome Y, for which only limited representation (∼18 Mb) was present in mRatBN7.2, which is discordant with Chromosome Y size expectations from the karyotype (Hamta et al. 2006). Four other chromosomes have increases in size of >10%: Chromosomes 3, 11, 12, and 19. In the case of Chromosomes 3, 11, and 12, the additional sequence is largely contributed near the proximal telomere. The acrosomal arms of these chromosomes are the location of the nucleolus-organizing regions of the rat genome, which are composed of multiple copies of ribosomal DNA genes (Tantravahi et al. 1979; Sasaki et al. 1986). These regions continue to pose a challenge to even the latest assembly methods (Rautiainen et al. 2023). We used SyRI software and the related plotsr visualization package (Goel et al. 2019; Goel and Schneeberger 2022) to analyze the syntenic relationships and structural differences between the new assembly and the mRatBN7.2. Figure 4 shows the synteny relationships among the five chromosomes in which the largest relative increases in size were observed. Supplemental Figure S2 provides similar images for the remaining chromosomes and also indicates inter-chromosomal exchanges between the assemblies. The sequence added to chromosomes in this new assembly generally occurs in contiguous blocks, often corresponding to the expected centromere locations (Hamta et al. 2006). In addition, small inversions are noted, as are several translocations and duplications. SyRI also provides a summary and the extent of various types of structural variation between mRatBN7.2 and the current assembly (Table 4). The “inversions,” “translocations,” and “duplications” noted in this table refer to segment-wise differences between two reference assemblies; they are not differences between individual animals.
Synteny analysis was performed with SyRI software to illustrate the shared regions of chromosomes with the largest percentage increase in size in the present assembly compared with mRatBN7.2. Chr 3, Chr 11, and Chr 12 are acrosomal chromosomes in which rDNA arrays are located in the acrosomal arms. The current assembly has greatly extended the chromosome in the region of these arms. Chr 19 shows a large insertion spanning >15 Mb that was not included in mRatBN7.2. Chr Y shows good synteny with mRatBN7.2 across the Y-specific coding regions of the chromosome. However, the more distal regions were not assembled in mRatBN7.2, undoubtedly reflecting the repetitive heterochromatic region of this chromosome.

Chromosome sizes in the GRCr8 assembly compared with those of the primary haplotype of mRatBN7.2, also indicating the difference in size (Δ) and % increase in size
| Chromosome | GRCr8 bp | mRatBN7.2 bp | Δ | % Added |
|---|---|---|---|---|
| Chr 1 | 270,518,180 | 260,522,016 | 9,996,164 | 3.84 |
| Chr 2 | 251,712,708 | 249,053,267 | 2,659,441 | 1.07 |
| Chr 3 | 189,428,310 | 169,034,231 | 20,394,079 | 12.07 |
| Chr 4 | 184,426,481 | 182,687,754 | 1,738,727 | 0.95 |
| Chr 5 | 172,190,305 | 166,875,058 | 5,315,247 | 3.19 |
| Chr 6 | 147,156,653 | 140,994,061 | 6,162,592 | 4.37 |
| Chr 7 | 137,014,596 | 135,012,528 | 2,002,068 | 1.48 |
| Chr 8 | 132,782,436 | 123,900,184 | 8,882,252 | 7.17 |
| Chr 9 | 121,768,150 | 114,175,309 | 7,592,841 | 6.65 |
| Chr 10 | 107,713,808 | 107,211,142 | 502,666 | 0.47 |
| Chr 11 | 99,753,367 | 86,241,447 | 13,511,920 | 15.67 |
| Chr 12 | 52,308,831 | 46,669,029 | 5,639,802 | 12.08 |
| Chr 13 | 109,350,286 | 106,807,694 | 2,542,592 | 2.38 |
| Chr 14 | 109,089,856 | 104,886,043 | 4,203,813 | 4.01 |
| Chr 15 | 108,192,169 | 101,769,107 | 6,423,062 | 6.31 |
| Chr 16 | 91,442,908 | 84,729,064 | 6,713,844 | 7.92 |
| Chr 17 | 93,515,804 | 86,533,673 | 6,982,131 | 8.07 |
| Chr 18 | 86,134,022 | 83,828,827 | 2,305,195 | 2.75 |
| Chr 19 | 74,246,245 | 57,337,602 | 16,908,643 | 29.49 |
| Chr 20 | 56,021,148 | 54,435,887 | 1,585,261 | 2.91 |
| Chr X | 157,758,123 | 152,453,651 | 5,304,472 | 3.48 |
| Chr Y | 59,846,084 | 18,315,841 | 41,530,243 | 226.74 |
| Total | 2,812,370,470 | 2,633,473,415 | 178,897,055 | 6.79 |
Structural and sequence annotations arising from comparison of GRCr8 and mRatBN7.2 assemblies using SyRI software
| Variation_type | Count | Length of GRCr8 (bp) | Length of mRatBN7.2 (bp) |
|---|---|---|---|
| Structural annotations | |||
| Syntenic regions | 115 | 2,643,106,557 | 2,615,590,561 |
| Inversions | 27 | 3,442,466 | 2,884,883 |
| Translocations | 79 | 3,577,812 | 3,580,878 |
| Duplications (GRCr8) | 79 | 1,581,927 | |
| Duplications (mRatBN7.2) | 21 | 155,272 | |
| Not aligned (GRCr8) | 295 | 160,909,795 | |
| Not aligned (mRatBN7.2) | 171 | 11,946,199 | |
| Sequence annotations | |||
| SNPs | 175,070 | 175,070 | 175,070 |
| Insertions | 196,324 | 1,527,995 | |
| Deletions | 87,274 | 4,603,406 | |
| Copy gains | 6 | 92,600 | |
| Copy losses | 8 | 957,763 | |
| Highly diverged | 1,981 | 45,639,987 | 21,500,783 |
| Tandem repeats | 4 | 12,524 | 12,857 |
We have examined the incorporation of new protein-coding genes into the assembly by using NCBI annotation data. These fall into two categories: 780 unmapped genes that could not be placed on the mRatBN7.2 assembly via whole-genome alignments but are localized to chromosomes in GRCr8, and 373 novel genes whose sequence is present in both assemblies but were not previously annotated on mRatBN7.2 by NCBI . A listing of these genes is provided in Supplemental Table 1. A majority of both categories of genes, including 76% of the unmapped and 60% of the novel genes, share homology with known genes. Many of the unmapped genes appear to be related to testis and sperm function and localize to repetitive regions of the genome in which expansion of these genes has taken place.
The pseudoautosomal regions of the sex chromosomes
When meiotic recombination between the X and Y Chromosomes occurs, it is limited to the pseudoautosomal region (PAR). In humans and other primates, the X and Y Chromosomes are (sub)metacentric. However, in murids (including rats and mice), the sex chromosomes are telocentric. Recent studies suggest that the PAR in murids has undergone progressive degeneration with loss of PAR genes either to autosomal locations or completely from the genome. We investigated in our assembly the chromosomal location of genes present in the human PAR. In the mouse (Mus musculus, Mmu), only two human PAR gene orthologs were identified in the mouse PAR (Maxeiner et al. 2020). Studies of a prior R. norvegicus genome assembly did not identify any PAR genes in the proximal ends of the sex chromosomes (Maxeiner et al. 2020). We examined the location of orthologs to genes located in the human PAR in our assembly. As is shown in Table 5, we confirmed the absence of prospective PAR genes in proximal sex chromosomes in our assembly, finding that such genes either were absent from the assembly or are located on autosomes. This suggests that recombination capacity mediated by PAR of the X and Y Chromosomes in humans may be absent in the rat and that the telomeric region of the rat sex chromosomes may have a function limited to chromosomal synapsis and chiasma formation.
Comparison of the location of 16 human Chr X and Chr Y pseudoautosomal region (PAR) genes with the corresponding mouse and rat genes
| Gene | Hsa RefSeq | Hsa position | Mmu RefSeq | Mmu position | Rn Seq | Rn position |
|---|---|---|---|---|---|---|
| PLCXD1 | NM_018390 | Chr X: 109,617–131,584 Chr Y: 108,952–130,733 | NM_207279.3 | Chr 5: 110,247,661–110,253,819 | XM_006249594.4 | Chr 12: 52,238,611–52,241,876 |
| GTPBP6 | NM_012227 | Chr X: 132,978–149,874 Chr Y: 132,127–148,960 | NM_145147.5 | Chr 5: 110,251,843–110,256,063 | NM_001135840.1 | Chr 12: 52,234,612–52,238,574 |
| PPP2R3B | NM_013239 | Chr X: 165,039–218,172 Chr Y: 168,543–224,805 | Absent | NM_001139492.1 | Chr 14: 103,483–110,740 | |
| SHOX | NM_001191546 | Chr X: 471,065–484,646 Chr Y: 484,200–497,713 | NM_001302357.1 | Chr 3: 66,879,056–66,889,104 | Absent | |
| CRLF2 | NM_022148 | Chr X: 1,035,704–1,058,464 Chr Y: 1,050,450–1,072,677 | NM_001310694.1 | Chr 5: 109,702,575–109,706,859 | NM_134465.2 | Chr 14: 118,930–123,637 |
| CSF2RA | NM_001161531 | Chr X: 1,116,112–1,155,181 Chr Y: 1,129,402–1,169,666 | NM_009970.2 | Chr 19: 61,212,840–61,216,856 | NM_001037660.1 | Chr 14: 113,073–118,247 |
| IL3RA | NM_002183 | Chr X: 1,187,106–1,226,335 Chr Y: 1,201,550–1,246,463 | NM_008369.2 | Chr 14: 8,114,270–8,123,357 | NM_139260.3 | Chr 12: 21,431,846–21,437,547 |
| SLC25A6 | NM_214418 | Chr X: 1,229,803–1,235,689 Chr Y: 1,250,365–1,255,930 | Absent | XM_039097301.1 | Chr 18: 70,846,358–70,847,597 | |
| ASMTL | NM_004192 | Chr X: 1,246,707–1,294,817 Chr Y: 1,266,964–1,315,887 | Absent | NM_001105915.2 | Chr 12: 21,429,098–21,431,849 | |
| P2RY8 | NM_178129 | Chr X: 1,304,499–1,338,509 Chr Y: 1,325,953–1,401,516 | Absent | Absent | ||
| AKAP17A | NM_005088 | Chr X: 1,393,022–1,403,887 Chr Y: 1,455,997–1,466,652 | NM_001418372.1 | Chr X: 169,098,201–169,105,547 | NM_001127246.2 | Chr 12: 21,424,191–21,428,933 |
| ASMT | NM_001171038 | Chr X: 1,416,353–1,441,907 Chr Y: 1,478,823–1,502,882 | NM_001308488.2 | Chr X: 169,106,356–169,111,844 | NM_144759.2 | Chr 12: 21,418,488,–21,423,338 |
| DHRSX | NM_145177 | Chr X: 1,829,953–2,113,734 Chr Y: 1,894,168–2,177,723 | NM_001033326 | Chr 4: 156,390,377–156,410,432 | NM_001105914.1 | Chr 12: 21,413,533–21,418,321 |
| ZBED1 | NM_004729 | Chr X: 2,099,191–2,113,734 Chr Y: 2,163,179–2,177,723 | Absent | Absent | ||
| CD99 | NM_002414 | Chr X: 2,304,268–2,354,297 Chr Y: 1,050,450–1,072,677 | NM_138309.3 | Chr X: 70,463,666–70,536,455 | NM_001100804.1 | Chr 20: 56,000,484–56,005,135 |
| XG | NM_001141919 | Chr X: 2,365,041–2,429,263 Chr Y: absent | Absent | Absent |
[i] The genes identified are all assigned to the human (Hsa) PAR, and most are present in both the X and Y Hsa PAR. In the mouse (Mmu), six of these genes are absent from the genome assembly (GRCm39), seven are on autosomes, and three are located on Chr X. In the GRCr8 assembly, four genes are absent from the assembly, whereas 12 are located on autosomes. Human data are from T2T assembly CHM 13v2.0. Mouse data are from GRCm39. Rat data are from BN-HiFi using annotation transfer from mRatBN7.2 (Liftoff).
Gene annotation
The mRatBN7.2 assembly was adopted by the GRC in November 2020 as the rat reference assembly, with the Rat Genome Database (RGD) as a GRC member for its ongoing curation and updates (Vedi et al. 2023). It has undergone thorough annotation using methodologies and data sets established at NCBI and Ensembl. GRCr8 has recently undergone annotation by the RefSeq group at NCBI. The annotation used about 10 billion short reads and about 35 million long reads and incorporates curated RefSeq transcripts (NM_ prefixes) for ∼80% of coding genes. We compared the recent June 2023 mRatBN7.2 NCBI annotation report (GCF_015227675.2-RS_2023_06) with the annotation report of GRCr8 (GCF_036323735.1-RS_2024_02). Notable advances are observed across most assessments, including the annotation of 1100 protein coding genes absent from the prior annotation. Table 6 provides a comparison and includes equivalent data for the current mouse reference assembly GRCm39 (NCBI Mus musculus annotation release 109). Although the mouse assembly annotation arises from a different data set of mouse RefSeq sequences, which includes a larger accumulation of cDNA and other sequencing data for mouse, it provides some indication of how the current rat reference annotation has advanced to a similar level as the current mouse reference. The prior rat reference assembly has more than 200 annotated genes that require additional curation to resolve discrepancies. Of these, 17 genes in the present assembly had the same problem as in mRatBN7.2, implying an origin in the RefSeq gene rather than the assembly. We used isoform sequencing (Iso-Seq) alignments to investigate 33 remaining genes with problematic concordance with the annotation gene set in GRCr8. Of these, 24 appeared to be problems arising in the annotation gene set, as the assembly was substantiated with concordant alignments. There were five genes that could not be investigated because no Iso-Seq alignments were present. Of the four remaining genes, one was not located in the assembly, two appear to reflect gaps in the assembly, and the final one was an MHC gene identified in a rat strain known to have a different MHC haplotype from the reference.
Comparison of NCBI annotation reports for mRatBN7.2, GRCr8, and GRCm39
| Feature | mRatBN7.2 | GRCr8 | GRCm39 |
|---|---|---|---|
| A. Genes, pseudogenes, mRNA's pseudo transcripts and CDS's annotated in the assemblies | |||
| Genes and pseudogenes | 42,054 | 47,357 | 50,561 |
| Protein-coding | 21,990 | 23,154 | 22,186 |
| Noncoding | 11,558 | 14,864 | 17,518 |
| Transcribed pseudogenes | 157 | 117 | 474 |
| Nontranscribed pseudogenes | 8005 | 8687 | 9869 |
| Genes with variants | 16,116 | 17,767 | 18,531 |
| Immunoglobulin/T cell receptor gene segments | 300 | 501 | 490 |
| Other | 44 | 34 | 24 |
| mRNAs | 73,402 | 85,576 | 92,486 |
| Fully supported | 72,688 | 84,885 | 92,206 |
| With >5% ab initio | 369 | 371 | 113 |
| Partial | 83 | 79 | 54 |
| With filled gap(s) | 0 | 0 | 0 |
| Known RefSeq (NM_) | 20,574 | 21,191 | 37,907 |
| Model RefSeq (XM_) | 52,828 | 64,385 | 54,579 |
| Noncoding RNAs | 24,927 | 30,129 | 38,629 |
| Fully supported | 21,377 | 26,261 | 35,041 |
| With >5% ab initio | 0 | 0 | 0 |
| Partial | 1 | 0 | 5 |
| With filled gap(s) | 0 | 0 | 0 |
| Known RefSeq (NR_) | 817 | 821 | 7443 |
| Model RefSeq (XR_) | 23,041 | 28,207 | 29,867 |
| Pseudotranscripts | 159 | 121 | 533 |
| Fully supported | 141 | 108 | 524 |
| With >5% ab initio | 0 | 0 | 0 |
| Partial | 0 | 0 | 0 |
| With filled gap(s) | 0 | 0 | 0 |
| Known RefSeq (NR_) | 43 | 43 | 461 |
| Model RefSeq (XR_) | 116 | 78 | 72 |
| CDSs | 73,715 | 86,077 | 92,989 |
| Fully supported | 72,688 | 84,885 | 92,206 |
| With >5% ab initio | 432 | 441 | 155 |
| Partial | 83 | 75 | 53 |
| With major correction(s) | 413 | 208 | 30 |
| Known RefSeq (NP_) | 20,574 | 21,191 | 37,920 |
| Model RefSeq (XP_) | 52,841 | 64,385 | 54,579 |
| B. featureCounts in the assemblies | |||
| Genes | 33,592 | 38,052 | 39,728 |
| All transcripts | 98,329 | 115,705 | 131,115 |
| mRNA | 73,402 | 85,576 | 92,486 |
| misc_RNA | 4637 | 4764 | 10,029 |
| miRNA | 796 | 796 | 2112 |
| tRNA | 737 | 771 | 422 |
| lncRNA | 16,266 | 21,024 | 23,611 |
| snoRNA | 1288 | 1564 | 1331 |
| snRNA | 1026 | 1013 | 999 |
| antisense_RNA | 1 | 1 | 9 |
| rRNA | 139 | 159 | 41 |
| telomerase_RNA | 1 | 1 | 64 |
| RNase_MRP_RNA | 1 | 1 | 1 |
| SRP_RNA | 1 | 1 | |
| Single-exon transcripts | 3200 | 3392 | 2941 |
| Coding transcripts (NM_/XM_) | 3179 | 3371 | 2710 |
| Noncoding transcripts (NR_/XR_) | 21 | 21 | 231 |
| Exons | 316,617 | 337,524 | 352,138 |
| In coding transcripts (NM_/XM_) | 272,844 | 284,365 | 285,412 |
| In noncoding transcripts (NR_/XR_) | 72,831 | 78,922 | 120,842 |
| Introns | 264,390 | 276,634 | 291,708 |
| In coding transcripts (NM_/XM_) | 233,700 | 240,878 | 243,819 |
| In noncoding transcripts (NR_/XR_) | 59,216 | 61,168 | 100,211 |
We used Liftoff software (Shumate and Salzberg 2021) to determine to what extent annotations (NCBI, UCSC, and Ensembl) of mRatBN7.2 could be transferred to the new assembly. This would provide an indication of the extent to which the new assembly had failed to incorporate gene coding regions into the assembly. Supplemental Table 2 indicates that only a handful of genes could not be transferred using this tool.
To gain further insight into the expressed genome, we used PacBio Iso-Seq data from multiple unique tissues of individuals from the same BN strain used for the assembly. These data are helpful to validate the NCBI annotation and provide further insight into potential novel isoforms. We have also evaluated the utility of these data to inform about gene expression from newly incorporated segments in this GRCr8 assembly. To examine this region further, we again used StainedGlass software to provide visualization of self-similarity across the region from Chr 19: 9 Mb–35 Mb. As can be seen in Figure 5, this revealed an extensive region of related repetitive sequences on Chromosome 19 from 14 Mb to 30 Mb.
Alignment of Iso-Seq data from several BN tissues obtained from the MCW suggested that the newly incorporated region on Chr 19 shows aligned transcript data across its extent. This is in contrast to newly incorporated centromeric regions in which no aligned transcripts were observed. To investigate further, we examined whether the included region might be composed of expanded genomic sequences with high similarity, such as might occur as the result of repeated segmental duplication. Self-to-self similarity visualization with StainedGlass reveals a highly duplicated structure that lacked the simple expansion observed in centromeric regions. This newly incorporated region of Chr 19 contains 276 previously unmapped RefSeq genes and 12 additional proposed genes from Gnomon annotation analysis.
Base-level assembly accuracy
Merqury analysis of GRCr8 provided a QV estimate of the assembly of 59.5 (Rhie et al. 2020). In our evaluation of mRatBN7.2, we found 129,000 variants shared by more than 156 inbred rats, from a data collection containing 163 inbred rats, which includes 12 BN/NHsdMcwi samples (de Jong et al. 2024).The most parsimonious explanation for the large number of variants shared by all rats, including BN/NHsdMcwi, the reference strain, is that these are not authentic variants unique to the Brown Norway rat genome, but rather result from PacBio CLR sequencing in the creation of the mRatBN7.2 assembly which is prone to sequencing errors. Because HiFi sequencing is much more accurate than CLR, we repeated this analysis, comparing variants shared by 42 rat strains, including several BN substrains, and discovered that only 550 variants were shared in common by most strains but divergent in our assembly. This provides a useful indication of the reduced inclusion of sequencing errors in the present assembly. Although much of the present assembly approaches the recently reported telomere-to-telomere assembly contiguity and completeness (Nurk et al. 2022; Rautiainen et al. 2023), additional new sequencing data, such as can be obtained using pore-based sequencing methods, will be required to generate an assembly in which remaining complex structural elements are resolved.
Discussion
We report an improved, accurate, and highly complete new reference assembly, GRCr8, for the rat, R. norvegicus. In comparison with GRCr8, the predecessor reference assembly, mRatBN7.2, comprises two components: a primary pseudohaploid assembly of 2.6375 Gb with 99.5% localized to chromosomes and an alternative (diploid) haplotype of 286.9 Mb. The mRatBN7.2 alternative haplotype is highly fragmented, containing 3156 scaffolds with a scaffold N50 of 1117 bases. The methodology used to create the mRatBN7.2 primary and alternate haplotypes reflects the expectation that the genome of the subject animal is outbred and contains widespread haplotypic variation that is diverted from the primary to the alternative assembly (Howe et al. 2021b). However, our k-mer analysis indicates a very highly inbred, if not completely inbred, genome.
Among the increased representation in this assembly, Chr Y is notably enlarged. Among the autosome, Chr 19 has incorporated a large new block of >15 Mb. We detected gene expression from this region by alignment of Iso-Seq transcripts. Expression was generally limited to transcripts obtained from testis, brain, ovary, stomach, skin, and/or thymus. Some of these alignments were shared across the tissues; however, the testis had much more unique transcript alignments in this locus. Exon structure and BLAST analysis suggested many of these reads were of related sequences, suggesting a genomic locus that had undergone extensive expansion. BLAST alignment indicated that many of the Iso-Seq transcripts in this region showed similarity to members of the Speer gene family, of which multiple copies have been annotated in the mouse genome (Spiess et al. 2003). Progeny sex ratio distortion has been observed in crosses between inbred mouse strains (Haines et al. 2021). Studies suggest a driver effect arising in the sex chromosomes, leading to gene amplification of genes expressed from the sex chromosome (Moretti et al. 2020). Among such genes are sex chromosome transcription factors, which may exert their sex ratio-distorting effects in part by acting on expression of autosomal genes. The target autosomal genes may also undergo ampliconic expansion (Herrmann et al. 1999). The abundant testis expression of multiple Speer genes from the Chr 19 locus in the rat suggests that this might be an ampliconic expansion that reflects drive arising from sex chromosome competition, as has been reported in the mouse (Moretti et al. 2020). Several other autosomal loci have been added to this assembly that also contain multiple copies of predominantly testis-expressed genes (e.g., Chr 15: 4.6–7.4 Mb; Chr 16: 20–25 Mb).
In common with the genomes of other rodents, the PAR of the rat genome indicates relocation of PAR genes to autosomes and loss of other PAR genes (Table 5). Erosion of the PAR has been proposed to result from invasion of repetitive sequences. Relocation appears to have occurred as a result of several distinct translocation events with Chr 12: 21 Mb being the autosomal site of five such translocated genes,
In conclusion, we report the generation of a new rat reference genome assembly, GRCr8, using contemporary methods that offers important advances from the current rat reference genome. It reveals increased completeness, correctness, and base-level accuracy and introduces features that reflect important underlying biological aspects of genomic function. Additional inbred rat genome assemblies are becoming available that reflect similar construction methods (Kalbfleisch et al. 2023) and will advance the utility of the rat as a genetic model of both basic genomic function as well as genetic studies of the genomic origin of phenotypes. These assemblies are available at RGD and provide the prospect of enhanced annotation in functional regions that have been unannotated or poorly annotated in the past, including highly duplicated regions that may not have been incorporated fully into prior rat assemblies but from which abundant gene expression occurs.
Methods
Whole-genome sequencing
The tissue sample used for the long-read sequencing was obtained from the liver of a male rat of the inbred BN strain aged 18 weeks. This subject animal was raised in a closed colony at the Medical College of Wisconsin with strain name BN/NHsdMcwi and was registered at the RGD under the identifier RGD_61498. High-molecular-weight (HMW) genomic DNA (gDNA) was isolated using a MagAttract HMW DNA Kit (Qiagen 67563) following the manufacturer's protocol. HiFi and CLR WGS SMRTbell libraries were generated as per manufacturer's instructions (Pacific Biosciences). SMRT sequencing was performed on the Sequel IIe instrument (Pacific Biosciences) using 2.0 chemistry and four SMRT cells. Following data collection, circular consensus sequencing (CCS) analyses were performed to generate highly accurate intramolecular consensus reads. Those CCS reads that demonstrated an intrinsic read quality of >Q20 (99%) were classified as “HiFi reads” and used for downstream assembly, resulting in 40-fold HiFi read coverage. We also used publicly available long-read CLR sequence data (European Nucleotide Archive accession ERR5310326 and ERR5310327) that was produced for the preceding rat reference genome assembly, mRatBN7.2 (Howe et al. 2021b), to provide gap filling information for the assembly. This sequence came from a male animal from the same strain and colony as used for the HiFi reads. Short-read sequences (150 bp end) with ∼50× coverage depth (TruSeq, Illumina HiSeq X) were also obtained from a BN male of the same strain and colony sequenced at the Medical College of Wisconsin (Sequence Read Archive IDs SRA: SRS17979924; SRA: SRS17979921; and SRA: SRS17979833). Short-read whole-genome sequencing data from 42 rat strains were obtained from NIH SRA (Supplemental Table 3) and were aligned to the assembly to validate base-level accuracy of the assembly.
Genome assembly
Assembly using HiFi reads employed HiCanu software (version 2.1.1), with an expected genome size set to 2.7 Gb (Nurk et al. 2020). The command line option -pacbio-hifi was used to specify the HiFi sequence FASTA files for assembly.
Assembly scaffolding
Two methods were employed to provide long-range information suitable for scaffolding the HiCanu-generated contigs. Optical genome mapping data were obtained from publicly available data (European Nucleotide Archive accession ERZ1741012) generated from the prior rat reference genome assembly. A hybrid assembly was created from the HiCanu contigs and a de novo optical map. Creation of the de novo map and its integration with the HiCanu contigs used Bionano Saphyr Solve software (v3.3) to generate hybrid scaffolds. Publicly available Arima Hi-C chromosome conformation capture data produced for the prior rat reference genome assembly (European Nucleotide Archive accession ERR5309023 and ERR5309024) were used to scaffold the hybrid scaffold assembly using SALSA2 software (Ghurye et al. 2019).
Chromosome builds, gap filling, and polishing
We evaluated and corrected scaffolds and integrated subchromosomal scaffolds into full-length chromosome builds using RagTag v2.1.0 software (Alonge et al. 2022). The existing NCBI rat reference assembly mRatBN7.2 (GCA_015227675.2) (Howe et al. 2021b) was used to template chromosome-level scaffolding. To increase joining gaps in the assembly, we used the “patch” module of RagTag in combination with the CLR data. The CLR reads were assembled with Canu software, and the resulting contigs were provided to RagTag for alignment-guided gap filling using minimap2 (Li 2018) to align the CLR reads to the unpatched assembly. Polishing of the assembly may reduce sequence inaccuracy arising from microsatellite and homopolymer repeat errors that can occur in HiFi-based assemblies and may improve accuracy in regions that were gap-filled with CLR-based contigs. Polishing can be done because the Brown Norway rat cannot homozygously differ from itself. In principle, any homozygous differences identified between its sequence data and the reference derived from them is an error and can be corrected. This was done with Pilon version 1.24 (Walker et al. 2014), where BN short-read WGS was mapped to the assembly. The memory required by Pilon was reduced by breaking the assembly up into individual scaffolds for analysis using the SAMtools faidx command (Li and Durbin 2009). Single-scaffold mapped data sets were extracted using SAMtools view, and each scaffold was processed through Pilon individually and aggregated back into a single assembly after polishing. It should be noted that the polishing step was run prior to the inclusion of the mitochondrial genome in the assembly. As such, reads that would have mapped to ChrM mapped to the nuclear-embedded mitochondrial (NUMT) DNA sequences instead. In some cases, this would have resulted in Pilon editing the nuclear genome to match alleles found in the mitochondrial genome. These artifacts along with the other homozygous differences that remain between the assembly and sequence data from the same strain after polishing are included as Supplemental Data tables in a tab-delineated text format (Supplemental Table 4).
Assembly annotation
Annotation of the assembly was performed by NCBI using standardized methods of the NCBI pipeline for reference genome annotation (Thibaud-Nissen et al. 2013). We also examined the ability of annotations from mRatBN7.2 to be transferred to the GRCr8 assembly using Liftoff software (Shumate and Salzberg 2021). New protein-coding genes in GRCr8 were identified using NCBI annotation files based on GeneIDs not found in the mRatBN7.2 annotation and were subdivided into “current-unmapped” and “current-novel” categories based on NCBI annotation comparison files, in which “unmapped” refers to genes with no comparable location on mRatBN7.2 found through assembly alignments and “novel” refers to genes with a comparable location but no gene was previously annotated.
Alignment of full-length BN/NHsdMcwi gene transcripts to the assembly
Samples were collected from 19 tissues (lung, spleen, liver, kidney, heart, brain, skin, stomach, ileum, colon, prostate, ovary, epididymal fat, urinary bladder, thymus, skeletal muscle, mixed vascular tissue, nucleated blood cells, and testis) from 18 week old BN/NHsdMcwi animals (one female from the same colony provided ovary; all other tissues came from the same male animal from which the HiFi sequencing was obtained). Tissues were placed in RNAlater until further use for long-read Iso-Seq preparation. Nucleated blood cells were purified by density centrifugation using SepMate reagents (Stem Cell Technologies) and transferred to RNAlater. RNA was extracted from ∼25 mg of tissue using the RNeasy Mini Kit (Qiagen), as recommended by the manufacturer. The quality and quantity of the RNA were assessed by a Bioanalyzer 2000 (Agilent) and Qubit fluorometry (Thermo Fisher Scientific), respectively. Barcoded Iso-Seq SMRTbell libraries were generated as recommended by the manufacturer (Pacific Biosciences) and sequenced using 2.1 chemistry and 30 h movies on the Sequel IIe system. Prior to sequencing, RNA samples were multiplexed in batches of four tissues, and each fourplex pool was sequenced using a single SMRTcell. Following data collection, HiFi reads were generated on the instrument and used as input into the Iso-Seq v3 workflow within the SMRT Link v10.0 online SMRT sequencing bioinformatics suite. Briefly, the Iso-Seq v3 pipeline trims barcodes, primers, and poly(A) tails; reorients transcripts; and filters out PCR concatemers, producing de novo predicted transcripts as “full-length nonconcatemer” (FLNC) reads that were used for downstream analyses. The BAM files generated by SMRT Link data processing of Iso-Seq data have been deposited at NCBI and are linked to the NCBI genome assembly project (NCBI PRJNA1027884).
The FLNC reads were converted to FASTA files and aligned to the genome assembly using minimap2. The resulting FLNC sam file was converted to BAM format using SAMtools, sorted, and indexed for viewing in Integrative Genomics Viewer (IGV) software (Robinson et al. 2011). The aligned Iso-Seq data have been used to investigate regions of the genome that were absent from the prior reference assembly to determine whether gene expression arises within these regions and to examine which tissues generate such reads.
Assembly evaluation
Completeness of the assembly was assessed using BUSCO, using compleasm software (Huang and Li 2023). We compared the new assembly with the prior assembly to identify regions of synteny or structural and base-level differences between them using SyRI software (Goel et al. 2019; Jiao and Schneeberger 2020; Goel and Schneeberger 2022). Base-level accuracy of the assembly was also investigated by comparison of the sharing of gene variants between other inbred rat strains and the GRCr8 assembly. Base-level accuracy of the assembly was also investigated by identifying genomic variants shared by all four BN/NHsdMcwi samples and at least 34 out of additional 38 inbred strains of rats. Short-read data from these samples were mapped against the GRCr8 assembly using BWA (Li 2013); variants were discovered using DeepVariant (Poplin et al. 2018) and jointly analyzed using GLnexus (Yun et al. 2021). Gor a list of SRA read records of these rat strain sequences, see Supplemental Table 3. We used StainedGlass software (Vollger et al. 2022) as a tool to identify centromeric structures within the assembly. This search was guided by prior studies of the rat karyotype (Hamta et al. 2006) as well as evidence from genome-to-genome alignment suggesting locations of newly incorporated sequences in the region of potential centromeres (see Fig. 4, Chr 19). We also searched the assembly to localize and measure the presence of telomeric hexameric repeats using the seqkt program (https://github.com/lh3/seqtk).
Data access
All sequencing data of the GRCr8 assembly generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1027884.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported in part by the National Center for Biotechnology Information of the National Library of Medicine (NLM) at the National Institutes of Health (NIH). This work was also supported by grant NIH–National Human Genome Research Institute R01HG011252 to P.A.D., M.L.S., and T.S.K., NIH–Office of the Director R24OD024617 to M.R.D. and A.E.K., and NIH–National Heart, Lung, and Blood Institute R01HL64541 to A.E.K. and M.R.D.
Author contributions: Original sample acquisition was by A.E.K. and M.R.D. Assembly and assembly evaluation were by K.L., J.C.B., J.M.D.W., K.H., J.L.C., and T.S.K. Original sample processing, sequence generation, and related data were by M.L.S., K.J.K., J.M.D.W., and K.H. Variant analysis was by H.C. Genome annotation was by P.M. and T.D.M. Project planning and oversight were by P.D., T.S.K., and M.L.S. Manuscript preparation was by P.A.D. Manuscript review and revision were by P.A.D., M.L.S., T.S.K., and T.D.M.
Notes
[4] Supplementary material [Supplemental material is available for this article.]
[5] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279292.124.
References
- ↵Alonge M, Lebeigle L, Kirsche M, Jenike K, Ou S, Aganezov S, Wang X, Lippman ZB, Schatz MC, Soyk S. 2022. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol 23: 258. 10.1186/s13059-022-02823-7
- ↵Astashyn A, Tvedte ES, Sweeney D, Sapojnikov V, Bouk N, Joukov V, Mozes E, Strope PK, Sylla PM, Wagner L, 2024. Rapid and sensitive detection of genome contamination at scale with FCS-GX. Genome Biol 25: 60. 10.1186/s13059-024-03198-7
- ↵Bao C, Zhu X, Mōller-Mara J, Li J, Dubroqua S, Erlich JC. 2023. The rat frontal orienting field dynamically encodes value for economic decisions under risk. Nat Neurosci 26: 1942–1952. 10.1038/s41593-023-01461-x
- ↵de Jong TV, Chen H, Brashear WA, Kochan KJ, Hillhouse AE, Zhu Y, Dhande IS, Hudson EA, Sumlut MH, Smith ML, 2022. mRatBN7.2: familiar and unfamiliar features of a new rat genome reference assembly. Physiol Genomics 54: 251–260. 10.1152/physiolgenomics.00017.2022
- ↵de Jong TV, Pan Y, Rastas P, Munro D, Tutaj M, Akil H, Benner C, Chen D, Chitre AS, Chow W, 2024. A revamped rat reference genome improves the discovery of genetic diversity in laboratory rats. Cell Genomics 4: 100527. 10.1016/j.xgen.2024.100527
- ↵Ghurye J, Rhie A, Walenz BP, Schmitt A, Selvaraj S, Pop M, Phillippy AM, Koren S. 2019. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Comput Biol 15: e1007273. 10.1371/journal.pcbi.1007273
- ↵Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, 2004. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493–521. 10.1038/nature02426
- ↵Goel M, Schneeberger K. 2022. plotsr: visualizing structural similarities and rearrangements between multiple genomes. Bioinformatics 38: 2922–2926. 10.1093/bioinformatics/btac196
- ↵Goel M, Sun H, Jiao W-B, Schneeberger K. 2019. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol 20: 277. 10.1186/s13059-019-1911-0
- ↵Haines BA, Barradale F, Dumont BL. 2021. Patterns and mechanisms of sex ratio distortion in the collaborative cross mouse mapping population. Genetics 219: iyab136. 10.1093/genetics/iyab136
- ↵Hamta A, Adamovic T, Samuelson E, Helou K, Behboudi A, Levan G. 2006. Chromosome ideograms of the laboratory rat (Rattus norvegicus) based on high-resolution banding, and anchoring of the cytogenetic map to the DNA sequence by FISH in sample chromosomes. Cytogenet Genome Res 115: 158–168. 10.1159/000095237
- ↵Hansen C, Spuhler K. 1984. Development of the National Institutes of Health genetically heterogeneous rat stock. Alcohol Clin Exp Res 8: 477–479. 10.1111/j.1530-0277.1984.tb05706.x
- ↵Herrmann BG, Koschorz B, Wertz K, McLaughlin KJ, Kispert A. 1999. A protein kinase encoded by the t complex responder gene causes non-mendelian inheritance. Nature 402: 141–146. 10.1038/45970
- ↵Howe K, Chow W, Collins J, Pelan S, Pointon D-L, Sims Y, Torrance J, Tracey A, Wood J. 2021a. Significantly improving the quality of genome assemblies through curation. GigaScience 10: giaa153. 10.1093/gigascience/giaa153
- ↵Howe K, Dwinell M, Shimoyama M, Corton C, Betteridge E, Dove A, Quail MA, Smith M, Saba L, Williams RW, 2021b. The genome sequence of the Norway rat, Rattus norvegicus Berkenhout 1769. Wellcome Open Res 6: 118. 10.12688/wellcomeopenres.16854.1
- ↵Huang N, Li H. 2023. compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics 39: btad595. 10.1093/bioinformatics/btad595
- ↵Jiao W-B, Schneeberger K. 2020. Chromosome-level assemblies of multiple Arabidopsis genomes reveal hotspots of rearrangements with altered evolutionary dynamics. Nat Commun 11: 989. 10.1038/s41467-020-14779-y
- ↵Kalbfleisch TS, Hussien AbouEl Ela NA, Li K, Brashear WA, Kochan KJ, Hillhouse AE, Zhu Y, Dhande IS, Kline EJ, Hudson EA, 2023. The assembled genome of the stroke-prone spontaneously hypertensive rat. Hypertension 80: 138–146. 10.1161/HYPERTENSIONAHA.122.20140
- ↵Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio.GN]. 10.48550/arXiv.1303.3997
- ↵Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191
- ↵Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324
- ↵Maxeiner S, Benseler F, Krasteva-Christ G, Brose N, Südhof TC. 2020. Evolution of the autism-associated neuroligin-4 gene reveals broad erosion of pseudoautosomal regions in rodents. Mol Biol Evol 37: 1243–1258. 10.1093/molbev/msaa014
- ↵Moretti C, Blanco M, Ialy-Radio C, Serrentino M-E, Gobé C, Friedman R, Battail C, Leduc M, Ward MA, Vaiman D, 2020. Battle of the sex chromosomes: Competition between X and Y chromosome-encoded proteins for partner interaction and chromatin occupancy drives multicopy gene expression and evolution in muroid rodents. Mol Biol Evol 37: 3453–3468. 10.1093/molbev/msaa175
- ↵Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, Grothe R, Miga KH, Eichler EE, Phillippy AM, Koren S. 2020. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res 30: 1291–1305. 10.1101/gr.263566.120
- ↵Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, 2022. The complete sequence of a human genome. Science 376: 44–53. 10.1126/science.abj6987
- ↵Poplin R, Chang P-C, Alexander D, Schwartz S, Colthurst T, Ku A, Newburger D, Dijamco J, Nguyen N, Afshar PT, 2018. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol 36: 983–987. 10.1038/nbt.4235
- ↵Pravenec M, Klír P, Kren V, Zicha J, Kunes J. 1989. An analysis of spontaneous hypertension in spontaneously hypertensive rats by means of new recombinant inbred strains. J Hypertens 7: 217–221.
- ↵Rautiainen M, Nurk S, Walenz BP, Logsdon GA, Porubsky D, Rhie A, Eichler EE, Phillippy AM, Koren S. 2023. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat Biotechnol 41: 1474–1482. 10.1038/s41587-023-01662-6
- ↵Rhie A, Walenz BP, Koren S, Phillippy AM. 2020. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21: 245. 10.1186/s13059-020-02134-9
- ↵Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754
- ↵Russell ES. 1985. A history of mouse genetics. Annu Rev Genet 19: 1–28. 10.1146/annurev.ge.19.120185.000245
- ↵Sasaki M, Nishida C, Kodama Y. 1986. Characterization of silver-stained nucleolus organizer regions (Ag-NORs) in 16 inbred strains of the Norway rat, Rattus norvegicus. Cytogenet Cell Genet 41: 83–88. 10.1159/000132208
- ↵Shumate A, Salzberg SL. 2021. Liftoff: accurate mapping of gene annotations. Bioinformatics 37: 1639–1643. 10.1093/bioinformatics/btaa1016
- ↵Solberg Woods LC, Palmer AA. 2019. Using heterogeneous stocks for fine-mapping genetically complex traits. Methods Mol Biol 2018: 233–247. 10.1007/978-1-4939-9581-3_11
- ↵Spiess A-N, Walther N, Müller N, Balvers M, Hansis C, Ivell R. 2003. SPEER: a new family of testis-specific genes from the mouse. Biol Reprod 68: 2044–2054. 10.1095/biolreprod.102.011593
- ↵Tabakoff B, Smith H, Vanderlinden LA, Hoffman PL, Saba LM. 2019. Networking in biology: the Hybrid Rat Diversity Panel. Methods Mol Biol 2018: 213–231. 10.1007/978-1-4939-9581-3_10
- ↵Tantravahi R, Miller DA, D'Ancona G, Croce CM, Miller OJ. 1979. Location of rRNA genes in three inbred strains of rat and suppression of rat rRNA activity in rat-human somatic cell hybrids. Exp Cell Res 119: 387–392. 10.1016/0014-4827(79)90368-9
- ↵Thibaud-Nissen F, Souvorov A, Murphy T, DiCuccio M, Kitts P. 2013. Eukaryotic genome annotation pipeline. In The NCBI Handbook, 2nd ed. National Center for Biotechnology Information, Bethesda, MD. https://www.ncbi.nlm.nih.gov/books/NBK169439/ [accessed April 29, 2022].
- ↵Uliano-Silva M, Ferreira JGRN, Krasheninnikova K, Darwin Tree of Life Consortium, Formenti G, Abueg L, Torrance J, Myers EW, Durbin R, Blaxter M, 2023. MitoHiFi: a python pipeline for mitochondrial genome assembly from PacBio high fidelity reads. BMC Bioinformatics 24: 288. 10.1186/s12859-023-05385-y
- ↵Vedi M, Smith JR, Thomas Hayman G, Tutaj M, Brodie K, C D, Pons JL, Demos WM, Gibson AC, Kaldunski ML, 2023. 2022 updates to the Rat Genome Database: a findable, accessible, interoperable, and reusable (FAIR) resource. Genetics 224: iyad042. 10.1093/genetics/iyad042
- ↵Voigt B, Kuramoto T, Mashimo T, Tsurumi T, Sasaki Y, Hokao R, Serikawa T. 2008. Evaluation of LEXF/FXLE rat recombinant inbred strains for genetic dissection of complex traits. Physiol Genomics 32: 335–342. 10.1152/physiolgenomics.00158.2007
- ↵Vollger MR, Kerpedjiev P, Phillippy AM, Eichler EE. 2022. StainedGlass: interactive visualization of massive tandem repeat structures with identity heatmaps. Bioinformatics 38: 2049–2051. 10.1093/bioinformatics/btac018
- ↵Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9: e112963. 10.1371/journal.pone.0112963
- ↵Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, Kriventseva EV, Zdobnov EM. 2018. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol 35: 543–548. 10.1093/molbev/msx319
- ↵Yun T, Li H, Chang P-C, Lin MF, Carroll A, McLean CY. 2021. Accurate, scalable cohort variant calls using DeepVariant and GLnexus. Bioinformatics 36: 5582–5589. 10.1093/bioinformatics/btaa1081
- ↵Zhou JL, de Guglielmo G, Ho AJ, Kallupi M, Pokhrel N, Li H-R, Chitre AS, Munro D, Mohammadi P, Carrette LLG, 2023. Single-nucleus genomics in outbred rats with divergent cocaine addiction-like behaviors reveals changes in amygdala GABAergic inhibition. Nat Neurosci 26: 1868–1879. 10.1038/s41593-023-01452-y