Abstract
The poly(A) signal, together with auxiliary elements, directs cleavage of a pre-mRNA and thus determines the 3′ end of the mature transcript. In many species, including humans, the poly(A) signal is an AAUAAA hexamer, but we recently found that the deeply branching eukaryote Giardia lamblia uses a distinct hexamer (AGURAA) and lacks any known auxiliary elements. Our discovery prompted us to explore the evolutionary dynamics of poly(A) signals and auxiliary elements in the eukaryotic kingdom. We use direct RNA sequencing to determine poly(A) signals for four protists within the Metamonada clade (which also contains G. lamblia) and two outgroup protists. These experiments reveal that the AAUAAA hexamer serves as the poly(A) signal in at least four different eukaryotic clades, indicating that it is likely the ancestral signal, whereas the unusual Giardia version is derived. We find that the use and relative strengths of auxiliary elements are also plastic; in fact, within Metamonada, species like G. lamblia make use of a previously unrecognized auxiliary element where nucleotides flanking the poly(A) signal itself specify genuine cleavage sites. Thus, despite the fundamental nature of pre-mRNA cleavage for the expression of all protein-coding genes, the motifs controlling this process are dynamic on evolutionary timescales, providing motivation for future biochemical and structural studies as well as new therapeutic angles to target eukaryotic pathogens.
Cleavage and polyadenylation (CPA) are key steps in eukaryotic mRNA maturation, specifying the 3′ end of the transcript and the addition of the poly(A) tail. Required for the proper expression of nearly all mRNAs, the site of pre-mRNA cleavage is determined by cis-regulatory RNA motifs, including the poly(A) (for polyadenylation) signal and auxiliary elements found upstream and downstream of the poly(A) signal. Decades of research have been spent defining these signals and their corresponding trans-acting factors in model systems like humans and yeast, but we know much less about the diversity and evolutionary dynamics throughout the eukaryotic tree of life.
The poly(A) signal is required for 3′ end processing. The most well-known poly(A) signal is the AAUAAA hexamer used in humans and other metazoans, originally discovered in the mid-1970s (Proudfoot and Brownlee 1976; Fitzgerald and Shenk 1981; Higgs et al. 1983; Montell et al. 1983). Elegant biochemical and structural studies have shown how this hexamer is recognized by a multiprotein complex known as the cleavage and polyadenylation specificity factor (CPSF) and directly bound by two components of the CPSF complex (CPSF30 and WDR33) with the remaining components facilitating cleavage (Mandel et al. 2006, 2008; Shi et al. 2009). Outside of metazoans, AAUAAA has been reported to be used in Arabidopsis and Schizosaccharomyces pombe (Graber et al. 1999b; Liu et al. 2017), while Saccharomyces cerevisiae prefer an A-rich poly(A) signal (Graber et al. 1999b; Liu et al. 2017). Some unicellular eukaryotes have been reported to use different and shorter poly(A) signal sequences; for instance, a UGUAA is used in the green algae Chlamydomonas reinhardtii and Oscreococcus lucimarinus (Shen et al. 2008; Zhao et al. 2019), while UAAA/GUAA tetramers or UAA trimers are used in red algae and diatoms, respectively (Zhao et al. 2019). We recently discovered that an assemblage A strain of the protist Giardia lamblia uses an unusual AGURAA poly(A) signal (Bilodeau et al. 2022). Thus, although the poly(A) signal is necessary for the correct expression of nearly all coding genes, there appears to be plasticity in the sequence itself, which is likely reflected in corresponding changes to the CSPF complex.
In most species studied to date, the poly(A) signal is rarely sufficient for cleavage and proper cleavage requires additional auxiliary elements (Sheets et al. 1990; Birse 1997). For instance, metazoans have two major auxiliary elements: an upstream U-rich motif and downstream U- and GU-rich motifs. The most canonical U-rich motif is a UGUA tetramer recognized by proteins in the Cleavage factor Im (CFIm) family (Brown and Gilmartin 2003; Venkataraman et al. 2005). U- and GU-rich sequences downstream from the cleavage site are recognized by cleavage stimulation factor proteins (CstF) (Beyer et al. 1997; Takagaki and Manley 1997; Zarudnaya et al. 2003). Auxiliary elements boost the assembly of the CPA machinery on the poly(A) signal and direct the endonuclease CPSF73 for cleavage of the nascent RNA (Takagaki and Manley 1997; Hu et al. 2005; Mandel et al. 2006; Sullivan et al. 2009). In yeast, UA-rich elements located ∼40 nt upstream are bound by the cleavage and polyadenylation factor 1B (CF1B), while U-rich elements surrounding the cleavage site are bound by the polyadenylation factor complex CPF (Guo and Sherman 1996; Graber et al. 1999a). Although less studied than the Amorphea clade (which contains humans and yeast) (Fig. 1A), auxiliary elements have also been defined in plants where far upstream elements are used although they lack a highly conserved consensus sequence (Wu et al. 1995; Rothnie 1996). GU-rich downstream sequences, similar to human ones, were also found in the parasitic protist Blastocystis hominis within the SAR clade (Li and Du 2014). We previously found that G. lamblia assemblage A does not use any known auxiliary element (Bilodeau et al. 2022), highlighting the potential diversity of cleavage site recognition within the eukaryotic tree.
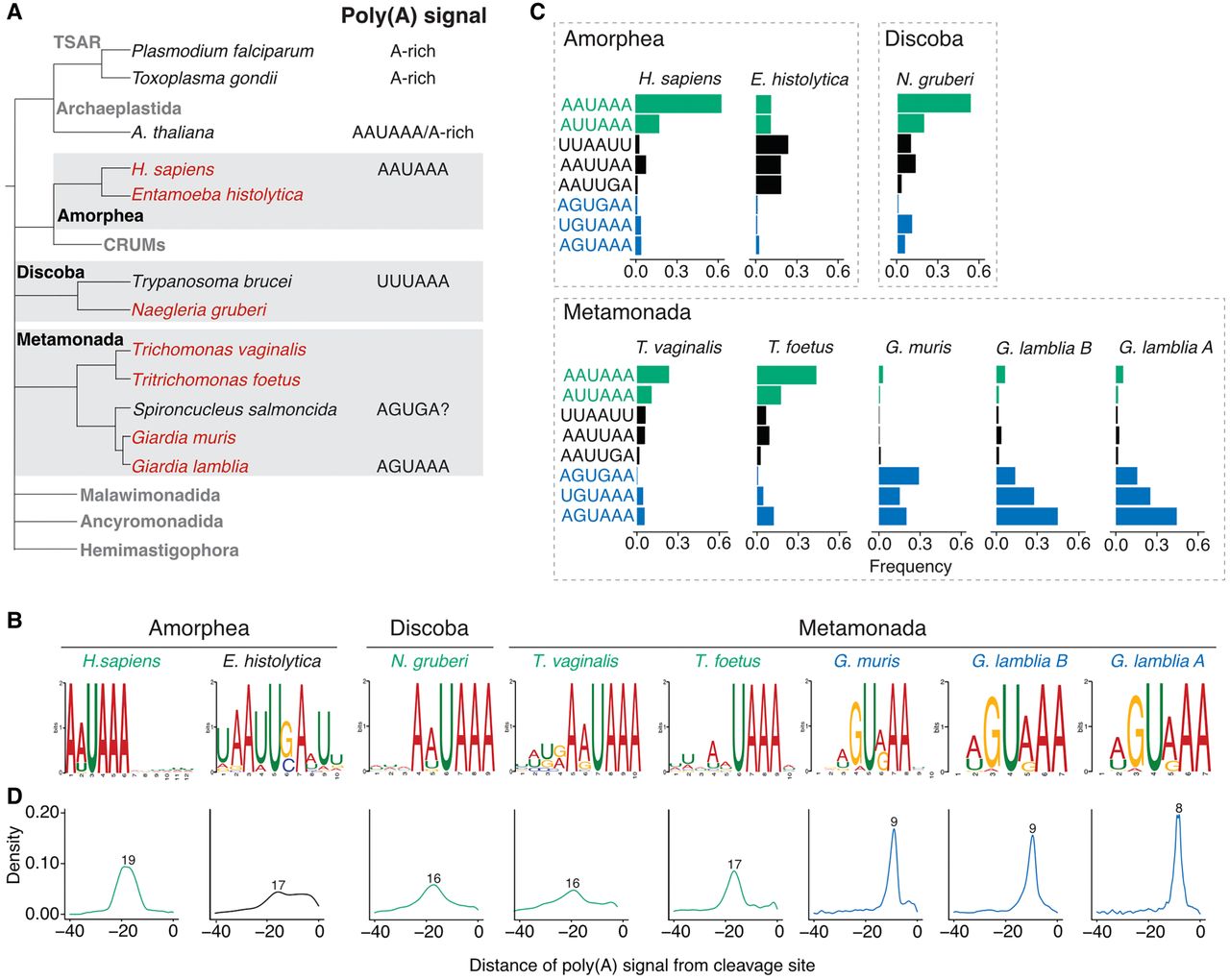
Characterization of poly(A) signals in diverse protists. (A) Simplified view of phylogenetic tree indicating evolutionary relationships between sequenced protist species (highlighted red) and other organisms. Known poly(A) signal sequences are shown next to the organism's name. (B) MEME analysis of sequence motifs enriched in the regions 40 nt upstream of the cleavage sites. Analysis was performed for all sequenced protist species and human annotated 3′-UTR sequences. Sequences representing previously described unusual AGURAA G. lamblia poly(A) signals were enriched within the Giardia genus. Sequences with enriched AWUAAA-like motifs are highlighted in green, AGURAA-like sequences in blue, and others in black. (C) Quantification of hexamers frequencies in annotated 3′ UTRs in the regions 40 nt upstream of the cleavage sites, based on the enrichment results from B. Color schemes the same as in B. (D) Distribution of the distances between poly(A) signals and cleavage sites. Poly(A) signals in the Giardia genus are positioned closer to the cleavage sites than in other organisms. Most common distance is shown above the plot. Color scheme is the same as in B.
A major limitation to our understanding of pre-mRNA cleavage is that knowledge of poly(A) signals, auxiliary motifs, and even 3′-UTR annotations are limited to an extremely small subset of eukaryotic species (the majority of which lie in the Amorphea clade) and to only a few of the ∼200,000 protist species. Thus, we know very little about the evolution and diversity of the motifs specifying pre-mRNA cleavage. For instance, a major gap is how poly(A) signals were identified in the eukaryotic common ancestor. One explanation is that the examples of AAUAAA in humans and possibly plants could reflect convergent evolution, possibly due to other sequence constraints. In such a case, perhaps the ancestral Metamonada species went down a different trajectory to use AGURAA. An alternative model is that AAUAAA is in fact the ancestral sequence, and the AGURAA hexamer emerged within the Metamonada clade, which would then have led to potentially fascinating—and unexplored—genomic adaptations. Similar questions surround the use of auxiliary elements, especially given the lack of any known ones in G. lamblia.
To shed light on these fundamental evolutionary questions, we set out to characterize the diversity and evolutionary dynamics of strategies used to recognize poly(A) sites in six protists, including four within the Metamonada clade, one within Amorphea, and one within the Discoba clade. We also aimed to determine how changes to poly(A) signals and auxiliary elements underlie genomic adaptations allowing Metamonada species, and G. lamblia in particular, to specify genuine, as opposed to premature, poly(A) signals.
Results
Annotation of 3′ UTRs across diverse eukaryotic species
We previously found that G. lamblia uses the distinct, but well-defined, AGURAA poly(A) signal (Bilodeau et al. 2022) rather than the AAUAAA signal used in many organisms, including humans (Chan et al. 2011). To understand how the poly(A) signal evolved in eukaryotes, we explored poly(A) signals in other eukaryotic species. Although our initial experiments with G. lamblia assemblage A used both short and long-read sequences to identify poly(A) signals (Bilodeau et al. 2022), we reasoned that direct RNA Oxford Nanopore Technology would be sufficient for the identification of poly(A) signals because the method primes from the 3′ end of a transcript and does not require high-quality genome annotations. Direct RNA sequencing also enables the determination of other features of interest, including 3′-UTR annotations and poly(A)-tail length.
We focused on several criteria to identify possible species to include in our survey: evolutionary distance to G. lamblia; availability of RNA samples; and existing annotations and other biological insights. Based on these criteria, we selected four protists from the Metamonada clade, including two related Giardia species, G. lamblia assemblage B (which is genetically diverse from the previously examined assemblage A strain) (Adam et al. 2013; Zajaczkowski et al. 2021), and Giardia muris, and two Trichomonadida species (Trichomonas vaginalis and Tritrichomonas foetus) from the separate Parabasalia phylum (Fig. 1A). We also selected two outgroup protists, Naegleria gruberi from the Discoba clade and Entamoeba histolytica from the Amorphea clade, which also contains well-studied organisms like Homo sapiens and S. cerevisiae.
We analyzed at least two biological replicates using direct RNA sequencing for each organism. Results from the replicates were highly correlated (Supplemental Fig. S1A), and the vast majority of reads contained untemplated adenosines, as expected (Supplemental Fig. S1C). In general, the median tail length was similar to that seen in all eukaryotic species, with the shortest occurring in N. gruberi (median length = 33). Nonetheless, even in this species, the poly(A) tail is sufficiently long to accommodate the predicted 26 nt footprint of poly(A)-binding protein (Supplemental Fig. S1B; Baer and Kornberg 1983). From a practical standpoint, tail lengths in these protists are long enough to introduce few biases during the standard oligo(dT)-enrichment step of RNA sequencing (except possibly for N. gruberi where care is warranted). For the rest of the analysis, we discarded reads with untemplated poly(A) tails shorter than 15 nt for N. gruberi and 30 nt for other protists, reasoning that these reads could represent decay intermediates rather than mature transcripts. Consistent with this interpretation, overall expression was lower for the discarded transcripts in comparison to the retained ones (Supplemental Fig. S1D).
Based on these criteria and minimum of 10 reads combined from all replicates, we were able to determine 3′ UTRs for 1409–5867 genes across the six species (Supplemental Fig. S1E). In many cases, our annotations are an improvement over previous annotations: For instance, before our study, no 3′ UTRs were annotated in T. foetus or in the assemblage B strain of G. lamblia. Our analysis identified 5867 and 2630 3′ UTRs, respectively, corresponding to 23% and 59% of all annotated genes for these organisms. We were also able to annotate 101–914 novel isoforms of previously annotated 3′ UTRs (Supplemental Table S1). In general, protist 3′ UTRs were substantially shorter than human 3′ UTRs (Supplemental Fig. S1F), and in some cases even shorter than those of G. lamblia, which has been previously noted for its short untranslated regions (Franzén et al. 2013; Bilodeau et al. 2022). For instance, the median length in E. histolytica and N. gruberi was 22 and 39 nt, respectively—meaning that the “functional” 3′-UTR sequence space for RNA-binding proteins seems to be minimal for many transcripts in several protists, especially once the ribosome shadow of ∼28 nt is considered (Yusupova et al. 2001; Takyar et al. 2005). Thus, it may be that ultrashort 3′ UTRs are more widespread than previously thought.
WGURAA polyadenylation signal is a derived trait in the Giardia genus
To determine the poly(A) signals for all seven protists, we next analyzed the sequence surrounding the cleavage sites of annotated 3′ UTRs. Because poly(A) signals are upstream of cleavage sites, we performed de novo motif discovery on the 40 nt upstream of the transcript end. Consistent with our previous work, both G. lamblia assemblage A and G. lamblia assemblage B showed enrichment of AGURAA-like sequences (Fig. 1B), as did G. muris. This extended analysis also revealed that UGUAAA was used to some extent in all Giardia species, leading us to redefine the consensus poly(A) signal as WGURAA (Fig. 1C, blue sequences). In other words, all Giardia species analyzed to date prefer “G,” not “A,” in the second position of the poly(A) signal.
A different picture emerged when we analyzed the other species. For the other Metamonada organisms (T. vaginalis and T. foetus), the most frequent hexamer was AAUAAA, followed by AUUAAA. Similarly, for N. gruberi in the Discoba clade, AAUAAA was again the top hexamer. This result is consistent with previous reports based on limited short-read data (Espinosa et al. 2002; Fuentes et al. 2012; Huang et al. 2013). Although E. histolytica showed more diverse poly(A) signal hexamer use, its most highly used sequences align with the previously identified AAWUDA sequence (Hon et al. 2013). Thus, together with previous reports in Arabidopsis thaliana and humans (Graber et al. 1999b), the AAUAAA hexamer has now been found in four clades (Archaeplastida, Amorphea, Discoba, and Metamonada), while the WGURAA hexamer appears to be restricted to Metamonada.
In most multicellular eukaryotes, canonical replication-dependent histone transcripts lack polyadenylation signals and poly(A) tails. In our analysis, we observed that most of the annotated histone transcripts were polyadenylated, which is consistent with the lack of an SLBP homolog in N. gruberi and G. lamblia (Yee et al. 2007; Dávila López and Samuelsson 2008). For all the protist species in our analysis, the poly(A) signal usage patterns for histone transcripts were similar to other transcripts (Supplemental Fig. S1G; Fig. 1C), which suggests common mechanisms of 3′ end formation.
To ensure that patterns of poly(A) signals usage we observed were not biased by transcripts that are polyadenylated but not transcribed by RNA polymerase II or processed by the nuclear 3′ end processing machinery, we repeated the analysis focusing on mRNAs encoding ribosomal proteins. These transcripts were highly conserved as well as highly expressed (Supplemental Fig. S1H). We observed the poly(A) signal usage pattern for transcripts encoding ribosomal proteins reflected the pattern for annotated 3′ UTRs (Supplemental Fig. S1I; Fig. 1C). Thus, the observed differences in poly(A) signal hexamers are unlikely to be explained by non-pol II transcripts with alternative 3′ end formation.
Additionally, we checked poly(A) signal usage in the transcripts removed from the analysis due to the short poly(A) tail, and it was consistent with the pattern observed in transcripts kept in the analysis (Supplemental Fig. 1J; Fig. 1C), except in the case of G. muris, when only ∼half of 3′ UTRs contain WGURAA or AWUAAA signals. However, this result can be explained because 29/61 of the transcripts removed in G. muris were rRNAs. This finding is in line with our previous observation that rRNA transcripts have short poly(A) tails likely processed by the TRAMP complex (Bilodeau et al. 2022).
We also investigated the positional preference of the poly(A) signals relative to the cleavage site because this distance was shown to be an important determinant of cleavage position (Chen et al. 1995). The poly(A) signals of E. histolytica, N. gruberi, T. foetus, and T. vaginalis were typically ∼15–20 nt upstream of the cleavage sites, which is similar to the distances reported for Metazoans (Fig. 1D; Kumar et al. 2019). The poly(A) signals in all three Giardia strains were much closer to the cleavage site and only 8–9 nt upstream.
We conclude that AWUAAA is the ancestral poly(A) signal, and that the use of “G” at the second position is a derived trait within, at least, the Giardia genus. This sequence preference coincides with positional preferences for cleavage sites, which may reflect changes in the underlying CPA machinery or other evolutionary constraints on the surrounding sequence elements.
“Dual use” stop codons are most frequent for WGURAA hexamers in Giardia species
One hallmark of organisms with highly compact genomes, like G. lamblia and Spironucleus salmoncida, are so-called “dual use” stop codons, where the stop codon itself also provides part of the poly(A) signal (Fig. 2A; Xu et al. 2014, 2020; Bilodeau et al. 2022). The shift in poly(A) signal increases the number of possible dual use stop codons from five to nine (Fig. 2A). To examine the potential extent of dual-use signals, we calculated the percentage of annotated protein-coding genes in which AAUAAA or WGURAA overlaps with a stop codon. We found that 7.4%, 12.8%, and 18.3% of them had potential dual-use signals for the WGURAA hexamer in G. lamblia A, G. lamblia B, and G. muris, respectively, but only 1%–2.1% for the AAUAAA hexamer (Fig. 2B).
“Dual use” signals in protists. (A) Switching the poly(A) signal from AAUAAA to WGURAA extends the number of possible “dual use” signals from 5 (left panel) to 9 (right panel). Poly(A) signal sequence is colored green (AAUAAA) or blue (WGURAA), stop codon sequence is highlighted gray. (B) Stop codons of 8%–20% of protein-coding genes in Giardia overlap with poly(A) signals. Almost all overlapping poly(A) signals are WGURAA. In contrast, AAUAAA makes up the majority of overlapping poly(A) signals for N. gruberi. (C) Length comparison between 3′ UTRs with and without “dual use” signals for G. lamblia A and N. gruberi. 3′ UTRs containing these signals are significantly shorter. Median length is shown below the plot.
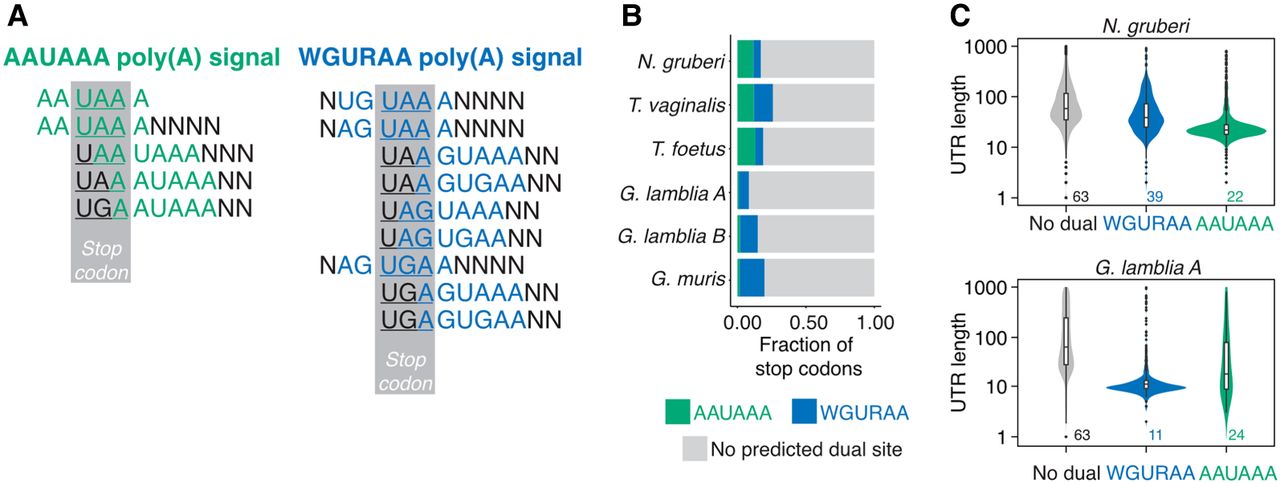
Given the short 3′-UTR lengths in non-Giardia protists such as N. gruberi, we examined the possibility that dual-use stop codons might play a role in these protists. (Due to the apparent degeneracy in poly(A) signal specification in E. histolytica, we did not include this species in this or further analysis.) Consistent with WGURAA not being a poly(A) signal in N. gruberi, and T. foetus, there was little overlap of this sequence with stop codons. In contrast, for AAUAAA, ∼11%–13% of stop codons overlapped with this signal in N. gruberi and T. foetus. Stop codons overlapped with AAUAAA and WGURAA to a similar extent in T. vaginalis (Fig. 2B). To assess, the potential of these putative dual-use stop codons to act as poly(A) signals, we examined the 3′-UTR lengths of the corresponding transcripts (Fig. 2C). As expected, and previously reported (Bilodeau et al. 2022), the median length of WGURAA dual-use sites in G. lamblia assemblage A was 11 nt, which is in line with the distance between poly(A) signals and cleavage sites in this species (Fig. 1D); for AAUAAA-containing sites, the median length was 24. The opposite picture emerged for N. gruberi, where the AAUAAA-containing sites had a median length of 22 nt compared to 39 nt for the WGURAA sites. These results indicate that N. gruberi also has dual-use stop codons, although they may be less common than in Giardia. In contrast, based on 3′-UTR lengths we saw limited evidence for dual-use sites in T. vaginalis and T. foetus. Thus, dual-use stop codons appear to be a common strategy to allow the production of ultrashort 3′ UTRs within some protist species.
Known auxiliary elements are absent in Metamonada
Poly(A) signals are necessary, but typically not sufficient to specify cleavage sites, and auxiliary elements are often used to help discriminate between “genuine” and “premature” sites. One important motif is the upstream UGUA motif, which is recognized in humans by CFIm. We had previously found little evidence for its use in G. lamblia assemblage A (Bilodeau et al. 2022) and wondered how evolution has shaped the use of the UGUA motif across eukaryotes. To look for evidence of its use, we compared the occurrence of the UGUA motif 20–50 nt upstream of the cleavage site to the shuffled versions (Fig. 3A; Supplemental Fig. S2). As expected, there was a clear signal in humans for this motif, and we also found evidence in N. gruberi for the presence of this motif. In contrast, we found no evidence for the UGUA motif in any Metamonada species, irrespective of poly(A) signal identity. Given the evidence of an analogous “far upstream element” in plants (Li and Hunt 1997), these data are consistent with a loss of the UGUA element early in Metamonada evolution.
Absence of metazoan-like auxiliary elements in protists. (A) Comparison of the UGUA versus its shuffled versions 20–50 nt upstream of the cleavage site in human versus protists. Each data point represents the total number of either UGUA tetramers (red) or its shuffled versions (gray) and the bar shows the mean count value for all compared combinations. We observed human-like enrichment only in N. gruberi. Statistical analysis is presented in Supplemental Figure S2. (B) Enrichment of GU and U-rich elements calculated in the region 40 nt downstream from the cleavage site in comparison to the region 40 nt upstream to the cleavage site. Each point on the box and whisker plot represents the log2 ratio from downstream versus upstream quantity of one of the CUGCCU, CUGGGG, CUGUGU, GUCUGU, GUGUCU, GUGUGU, UGUCUC, UGUCUG, UGUGUG, UGUUUU, UUAUUU, UUUCUU, UUUUUU sequences. Statistical significance was calculated in comparison to human enrichment using a one-sided Wilcoxon rank sum test.
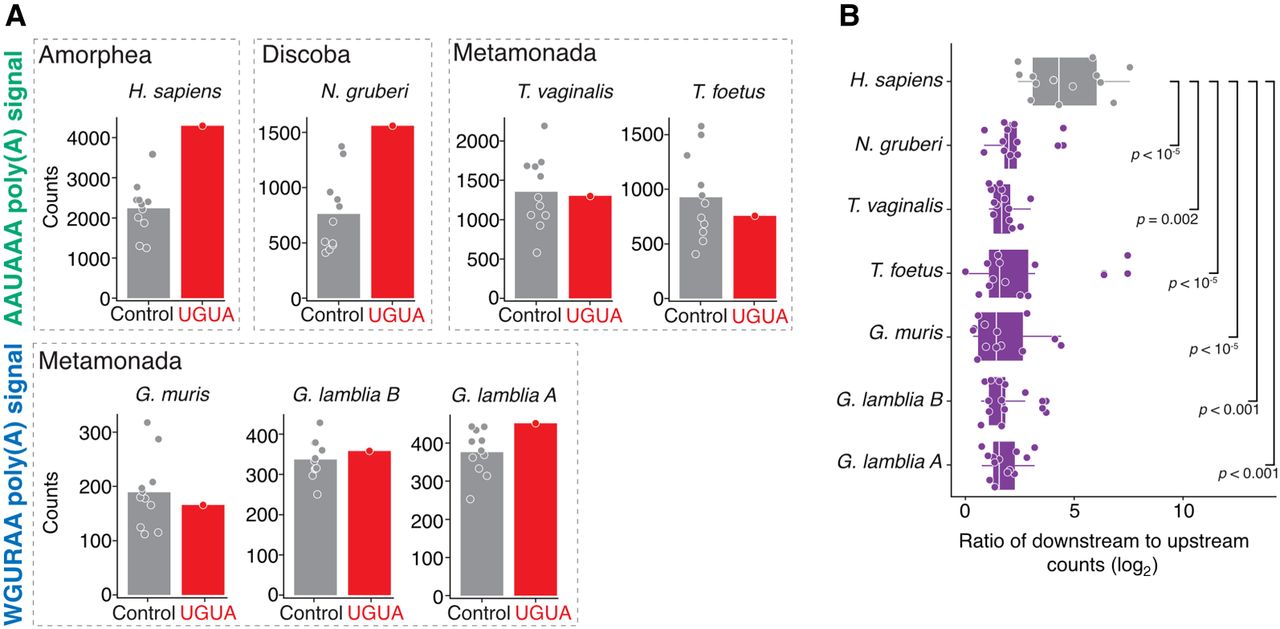
Because downstream GU- and U-rich elements also help specify poly(A) sites in metazoans, we next examined these motifs among the protists, comparing their occurrence 40 nt downstream from the cleavage site with those 40 nt upstream (as a control set). Using the ratio in humans as a benchmark, we found that all protist species in our analysis exhibited significantly less downstream GU- and U-rich positional bias (Fig. 3B). Taken together, our results highlight a substantial plasticity in the role of auxiliary elements within Eukarya; most notably, those in the Metamonada clade lack known auxiliary elements, which suggests there may be an alternative mechanism(s) of poly(A) signals recognition and discrimination in this clade.
Nucleotides flanking the poly(A) signal are crucial for proper discrimination in G. lamblia
Premature cleavage has the potential to result in reduced or absent gene expression due to truncated open reading frames and subsequent repression by surveillance pathways. Our results thus far posed a riddle: How did a new poly(A) signal evolve without triggering premature cleavage at sites that previously would not have been recognized, especially given the apparent absence of other cis-elements to discriminate between true and premature sites? We hypothesized that one mechanism might be the reduced occurrence of the signal itself within open reading frames. Because coding region sequences are also shaped by amino acid biases, we first compared the occurrences of the AGU–AAA dicodons (which encode serine–lysine and could also serve as a poly(A) signal) with the eleven other synonymous codon combinations (Fig. 4A). The AGU-AAA dicodon was depleted relative to the other combinations in the Giardia species but not in T. vaginalis or T. foetus, which use the ancestral signal. We observed similar results with the other minor poly(A) signals (Supplemental Fig. S3A). Such depletions were not observed when we reversed the codons (e.g., AAA-AGU) (Supplemental Fig. S3A). Thus, the WGURAA poly(A) signal is depleted specifically in the coding regions of Giardia species in a manner that cannot be explained by amino acid bias. This depletion is especially pronounced in G. muris, where there are only 199 occurrences of the WGURAA hexamer in any frame (Fig. 4B), roughly 15× less than in G. lamblia A and G. lamblia B.
Importance of 3′ adjacent nucleotide of the WGURAA hexamer for distinguishing poly(A) signals from open reading frame hexamers. (A) Occurrence of AGU AAA dicodon in comparison to other Ser-Lys encoding dicodons in coding sequences within Metamonada clade. Dicodon occurrence was calculated only in frame. Each point represents one dicodon combination and the bar shows the mean count value for all compared combinations. AGU AAA was shown to be depleted in the Giardia genus, but not in other species. Data for AAA AGU, UGU AAA, and AGU GAA dicodons are shown in Supplemental Figure S3A. (B) WGURAA hexamer occurrence in the coding sequences of Giardia species. Occurrence was calculated independent of the reading frame. (C) Schematic representation of the machine learning approach to distinguish WGURAA sequences in 3′ UTRs versus coding sequences. WGURAA sequences were extracted from coding sequences and 3′ UTRs and together with 37 flanking nucleotides from both sides (80-mer) put into a gapped k-mer support vector machine classifier, which performed sequence classification and k-mers scoring. (D) Variance explained by the linear model applied to WGURAA-containing k-mers scores from gkmSVM classifier by the full model, upstream nucleotide, poly(A) signal, and downstream nucleotide in G. lamblia. Explained variance was measured as an adjusted R2 value. Data for G. lamblia B are shown in Supplemental Figure S3C. (E) Beta-coefficient values from the linear model applied to WGURAA-containing k-mers scores from gkmSVM classifier, corresponding to the upstream nucleotide, poly(A) signal, and downstream nucleotide in G. lamblia. Data for G. lamblia B are shown in Supplemental Figure S3B. (F) Example of G. lamblia A gene GL50803_1890 where a hexamer in the coding sequence was misclassified by gkmSVM. Premature cleavage after the hexamer inside the coding sequences indicated as coverage drop was observed. Similar example from G. lamblia B is shown in Supplemental Figure S3D.
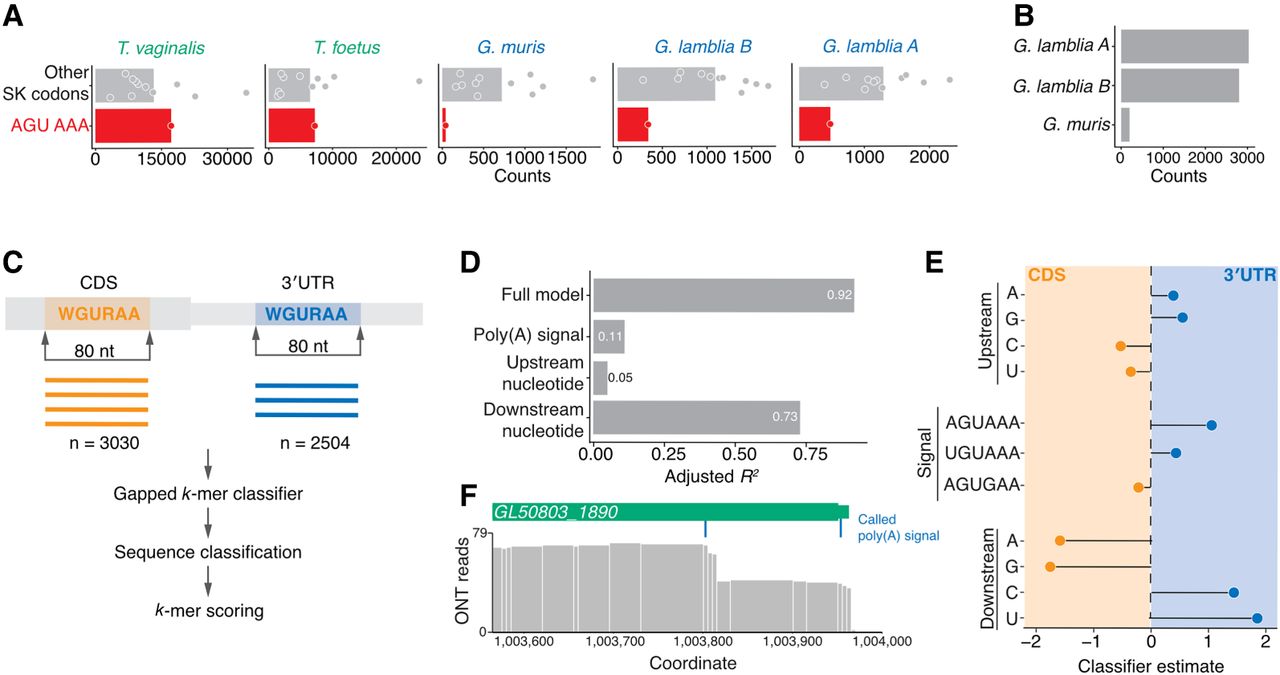
Nonetheless, in G. lamblia A and G. lamblia B, we still found thousands of examples of the poly(A) signal within coding regions (Fig. 4B), suggesting that mechanisms exist that allow discrimination of the correct poly(A) signal to avoid premature CPA in these situations. Consequently, we hypothesized that unique but heretofore unknown cis-elements must surround the true poly(A) signals. To identify such elements, we focused first on G. lamblia and used a gapped k-mer support vector machine (gkmSVM) (Ghandi et al. 2014, 2016) to classify the 80 nt regions surrounding WGURAA signals in the coding sequence (i.e., “premature”) and 3′ UTRs (i.e., “genuine”) (Fig. 4C).
Testing several k-mer lengths revealed minimal increase in classifier performance beyond 8 nt with the gkmSVM model performing exceedingly well (as judged by an F1 score exceeding 0.9) (Supplemental Table S2). Given that the 8 nt model only minimally exceeds the poly(A) signal length, we wondered whether nucleotides directly surrounding the poly(A) signal might be important for its recognition. We extracted gkmSVM scores for every 8-mer containing WGURAA sequences and applied a linear model to determine the extent to which the upstream nucleotide, downstream nucleotide, and poly(A) signal sequence could explain the variation in 8-mer scores. Specifically, pyrimidines were enriched downstream from the 3′-UTR classified sites, and AGUAAA was more associated with 3′ UTRs, consistent with a model that this hexamer is “stronger” than AGUGAA (Fig. 4D; Supplemental Fig. S3B).
These three features—the upstream and downstream nucleotides, and poly(A) signal—explained at least 90% of the variance in 8-mer scores for both G. lamblia A and G. lamblia B. When the contribution of each feature was determined individually, the downstream nucleotide had the largest contribution, explaining more than 70% of the variance, followed by the poly(A) signal identity (e.g., AGUAAA vs. AGUGAA, 11% of the variance) and then the upstream nucleotide (5% of the variance) (Fig. 4E; Supplemental Fig. 3C). Thus, the flanking nucleotides, especially the nucleotides directly downstream from the poly(A) signal, are important for distinguishing genuine poly(A) signals from premature ones.
To explore the possibility that additional, but as-yet-unknown, auxiliary elements might support poly(A) signal discrimination and detect putative regulatory elements shorter than 5 nt (the minimal k-mer length for gkmSVM), we generated de novo position weight matrices (PWMs) by systematically merging the most predictive k-mers from trained gkmSVM models (Ghandi et al. 2014). The top putative regulatory elements still overlapped with WGURAA and therefore contained at least a partial poly(A) signal and flanking nucleotide (Supplemental Fig. S3D,E), but outside these features, the sequences were highly degenerate. Thus, it appears that little beyond the 8 nt element containing the poly(A) signal helps differentiate between premature and true cleavage sites.
Despite the high performance of gkmSVM models, some coding sequences were misclassified as 3′ UTRs. These exceptions were especially interesting because they contained 3′-UTR-like features. We wondered if these might be examples of premature cleavage and visually inspected them. Of the 23 G. lamblia potentially false positive genes, 14 genes, including GL50803_1890 (Fig. 4F), contained reads consistent with premature cleavage events. The remaining sites were due to either incorrect gene annotations (seven cases) or classifier errors (two cases). Similarly, of the eight G. lamblia B false positives, five showed evidence of premature cleavage events (Supplemental Fig. S3F). Given that such premature cleavage is likely to reduce protein expression, it was interesting to note that three of these sites were within duplicated genes (two in G. lamblia A; one in G. lamblia B), and at least one copy of the duplicated genes lacked WGURAA hexamer (Supplemental Fig. S3G,H), providing a potential explanation for how the consequences of a functional premature cleavage site may be minimized within G. lamblia.
Majority of coding region WGURAA hexamers are recognized as poly(A) signals in G. muris
Given that G. muris contains very few WGURAA hexamers, we wondered about the extent to which flanking nucleotides helped discriminate between poly(A) signals in this organism. We again used the gkmSVM classifier and found the G. muris model also had high performance with an F1 score of 0.99. In contrast to our results with G. lamblia, however, the linear model only explained 51% of the variance in 8-mer scores (Fig. 5A; Supplemental Fig. S4A). Given the low performance of the linear model, we asked if dependencies between flanking nucleotides and poly(A) signal sequences may be important. Indeed, by including interactions between terms, the linear model now explained 85% of the variance (Fig. 5A).
Functional poly(A) signals in the majority of WGURAA hexamers in coding sequences in G. muris. (A) Variance explained by the linear model applied to WGURAA-containing k-mers scores from gkmSVM classifier by the full model, upstream nucleotide, poly(A) signal, and downstream nucleotide in G. muris. Explained variance was measured as an adjusted R2 value. The green bar represents a full linear model without interactions, blue—with interactions, gray—components of the model with interactions. Beta-coefficient values are found in Supplemental Figure S4A. (B) Influence of upstream and downstream nucleotide interaction to AGUGAA hexamers classification. Upstream nucleotide is color-coded at the top of each barplot. Data for AGUAAA and UGUAAA hexamers are shown in Supplemental Figure S4B and C, respectively. (C) Quantification of premature cleavage events in G. muris coding sequences by hexamer identity. Among 199 WGURAA hexamers in G. muris coding sequences, 106 had sufficient read support and 91 were cleaved.
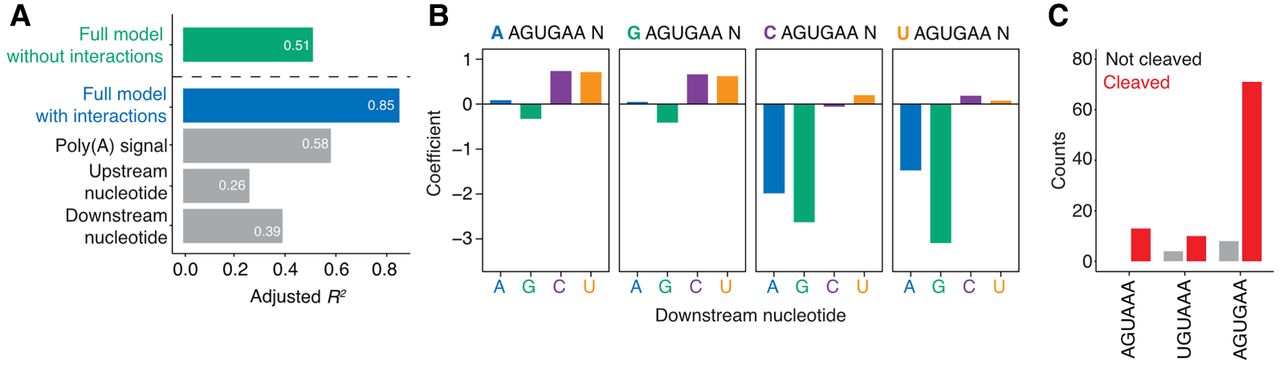
The G. muris model differed in several important ways from G. lamblia. First, although the poly(A) signal and flanking nucleotides each contributed to the model, the poly(A) signal was now the dominant feature with AGUGAA indicating a coding region site, but AGUAAA and UGUAAA occurring very infrequently in this region. As in G. lamblia, downstream purines were associated with coding region sites in G. muris, but these effects were strongest when coupled with upstream pyrimidines (Fig. 5B; Supplemental Fig. S4B,C). These findings indicate that in G. muris, the full octameric context is required to distinguish between premature and true cleavage sites.
Although the classifier was able to correctly recognize all coding sequences containing WGURAA hexamers, we wondered about the extent of premature cleavage in G. muris. We had sufficient read support for 106 genes and observed premature cleavage in 86% cases (Fig. 5C). We conclude that despite flanking sequence differences distinguishing WGURAA hexamers within coding sequences from those within 3′ UTRs, a substantial fraction has remained as functional poly(A) signals.
Flanking nucleotides play a role throughout the Metamonada clade
Having found evidence for a role of flanking nucleotides within Giardia species, we explored if the flanking nucleotides might have coevolved with the derived poly(A) signal, perhaps enabling adaptation to the change in the recognized hexamer. As before, we trained gkmSVM classifiers for T. foetus, T. vaginalis, and the outgroup species of N. gruberi. The classifier showed high performance for all three with the highest being for N. gruberi (F1 = 0.9), and lower for T. foetus and T. vaginalis (F1 = 0.78 for both).
The linear models for T. foetus and T. vaginalis performed less well than for Giardia (57% and 55%, respectively), but they still revealed the relative importance of poly(A) signals and flanking nucleotides. Indeed, contrary to our original hypothesis, the upstream and downstream flanking nucleotides contributed to model performance for both T. vaginalis and T. foetus. In other words, despite these species using the ancestral poly(A) signal, flanking nucleotides still play a role in poly(A) site discrimination (Fig. 6A,B). Incorporating dependencies between poly(A) signals and flanking nucleotides in T. foetus and T. vaginalis did not improve model performance (Supplemental Table S2). Thus, the three major elements of poly(A) signal and flanking nucleotides contribute substantially to poly(A) site recognition in all five Metamonada species analyzed. In the case of T. foetus and T. vaginalis, we suspect they utilize additional unknown mechanism(s) given the frequent occurrence of AAUAAA hexamers in the coding sequences.
Flanking nucleotides play a role in poly(A) signal recognition in other Metamonada, but not N. gruberi. (A) Variance explained by the linear model applied to AWUAAA-containing k-mers scores from gkmSVM classifier. In the outgroup organism N. gruberi, signal sequence is the main determinant, whereas in T. vaginalis and T. foetus from Metamonada upstream nucleotides have substantial contributions. (B) Beta-coefficient values from the linear model form A. (C) AAUAAA and AUUAAA hexamer occurrence in the coding sequences of N. gruberi. Occurrence was calculated independent of the reading frame. (D,E) Auxiliary elements help specify genuine AAUAAA poly(A) signals in N. gruberi, putative upstream (D) and downstream (E) motifs and their quantifications around AAUAAA poly(A) signals.
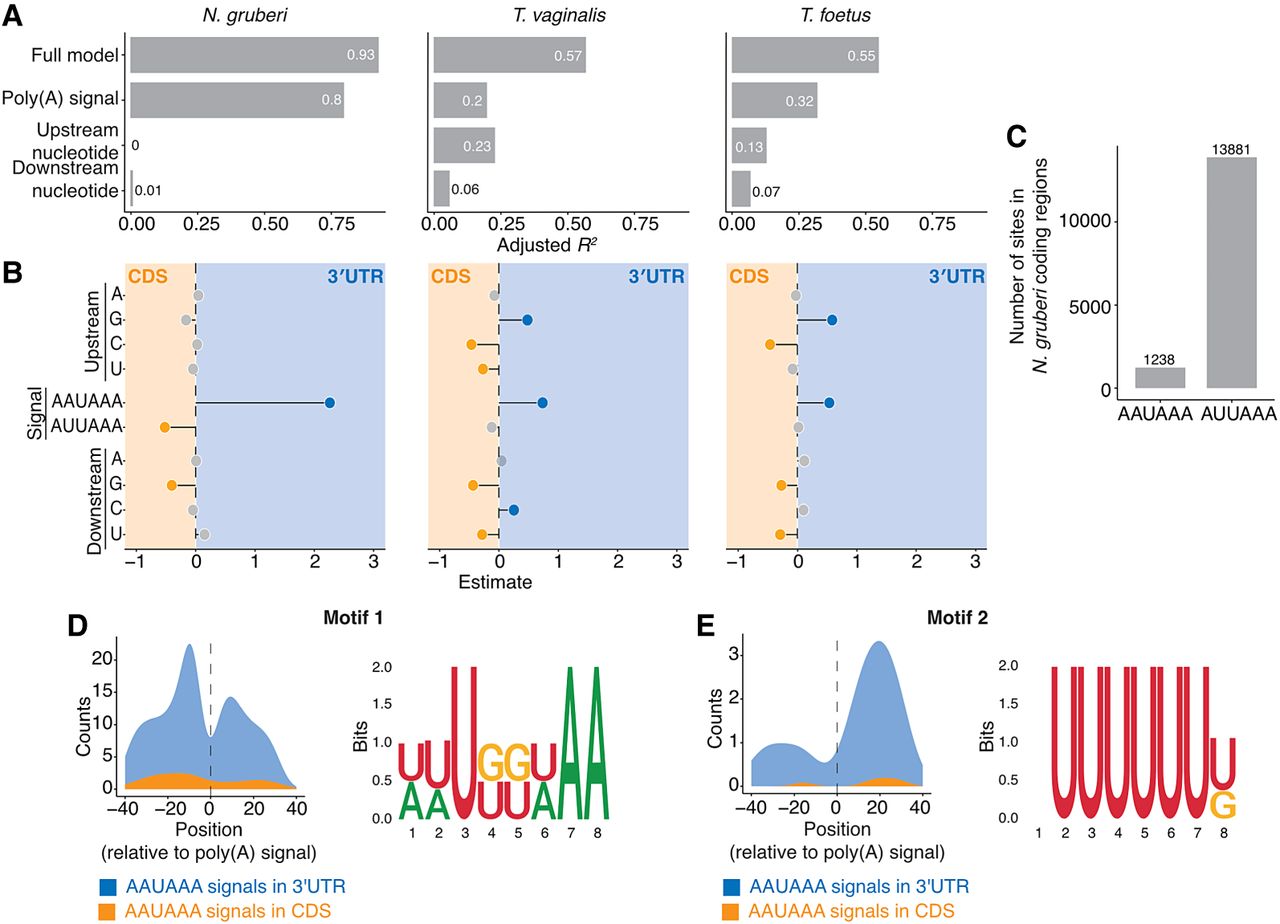
In N. gruberi, the linear model for 8-mer scores explained 93% of the observed variance with most of the contribution coming from the poly(A) signal, while the flanking nucleotides explained none of the variance. The strongest differentiator appeared to be AAUAAA in 3′-UTR sites, while AUUAAA occurred in coding regions, an observation which is consistent with AAUAAA acting as a stronger poly(A) signal. However, we still found 1238 instances of AAUAAA hexamers in coding sequences (Fig. 6C). We were curious to understand if and how proper poly(A) signal discrimination occurred for transcripts containing the AAUAAA signal in both coding sequences and 3′ UTRs and retrained the classifier on these sequences. This model also had very high performance (0.96), which suggested the presence of other auxiliary elements since the poly(A) signal for this restricted set was the same between coding sequences and 3′ UTRs. As with the full set, flanking nucleotides still made almost no contribution. To figure out the identity of the auxiliary elements, we clustered the top 50 8-mers with the highest gkmSVM scores. One of the sequences we identified contained a UGUA tetranucleotide and was preferentially located upstream of the poly(A) signal (Fig. 6D). The other sequence was U-rich and preferentially located downstream from the poly(A) signal in 3′ UTRs but not in coding sequences (Fig. 6E). Thus, N. gruberi uses UGUA as an upstream element, which is consistent with its enrichment (Fig. 3A). Our earlier analysis showed that, en masse, U- and GU-rich sequences had a lower downstream to upstream ratio in N. gruberi compared to humans (Fig. 3B). Together, these data indicate that N. gruberi chiefly use the identity of the poly(A) signal for proper discrimination, which is supplemented by the same primary upstream and downstream auxiliary elements used in metazoans.
Discussion
In this study, we used long-read direct RNA sequencing to map cleavage and poly(A) sites in six protist species to understand the evolution of poly(A) signals and auxiliary regulatory elements. We generated high-resolution annotation for 18,852 3′ UTRs across six protist species, which will be a valuable resource for the scientific community. We previously found that G. lamblia A uses the unusual poly(A) signal, WGURAA (Bilodeau et al. 2022). Here, we confirmed that finding and extended it by identifying the same poly(A) signal in other species within the Giardia genus. However, other Metamonada protists (T. vaginalis, T. foetus) as well as those in Discoba (N. gruberi) use the AAUAAA poly(A) signal, now providing evidence of this signal in four eukaryotic clades. Based on parsimony, we conclude that AAUAAA represents the ancestral poly(A) signal and that WGURAA is a derived trait within the Giardia genus. This result is in agreement with previous study suggesting that AAUAAA or its 3′ part UAAA is an ancestral poly(A) signal (Zhao et al. 2019). Because the fish pathogen Spironucleus salmonicida has been reported to use an AGUGA poly(A) signal (Xu et al. 2014), we propose that the shift from the ancestral signal occurred in the Diplomonada order (Fig. 7), although more precise evolutionary placement will require analysis of additional species.
Putative evolutionary events that shaped the evolutionary dynamics of poly(A) signal recognition. Multiple evolutionary events have shaped poly(A) signal recognition throughout eukaryotes, including involvement of flanking nucleotides, loss of importance for auxiliary elements, and change in the sequence of the poly(A) signal itself. To identify more precise placement of the hexamer change, additional annotations from more organisms are required.
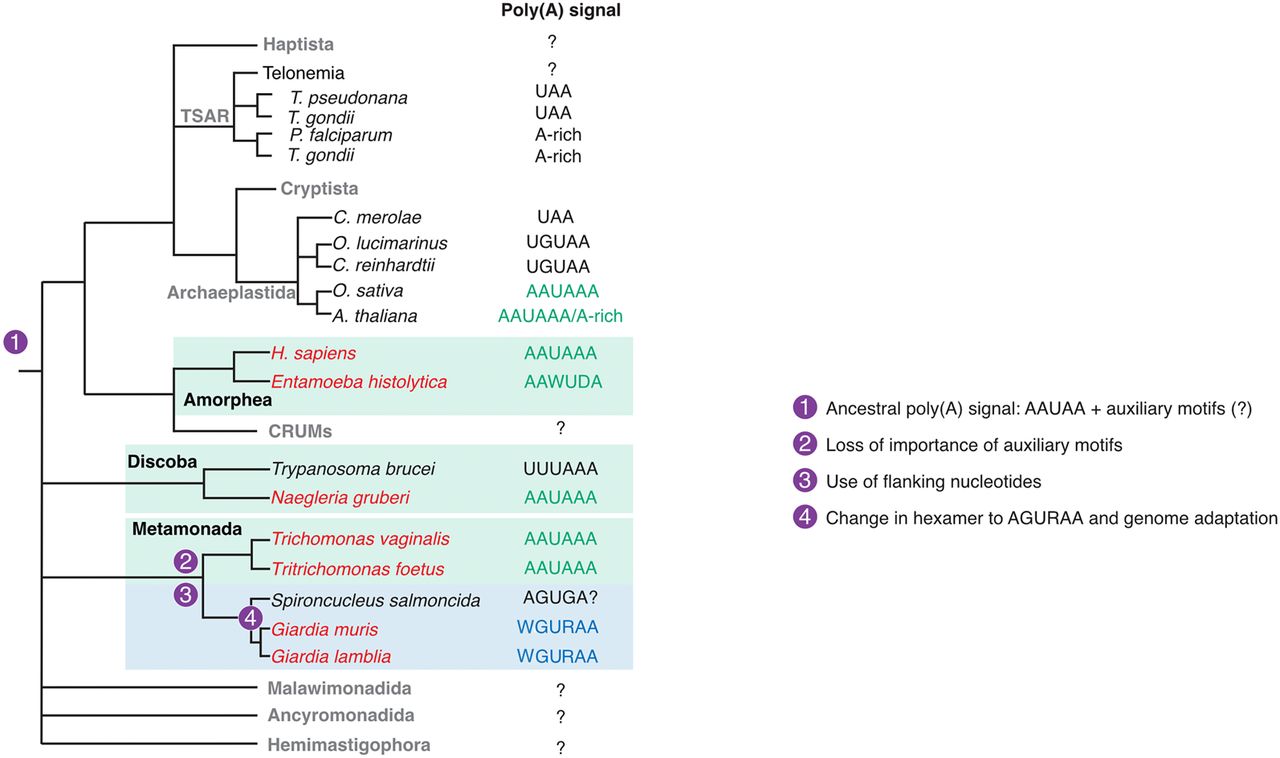
Previous work in several species had focused on the known auxiliary elements of the upstream UGUA and downstream U- and GU-rich elements. In the case of N. gruberi, UGUA motifs are enriched upstream of poly(A) signals, and U-rich sequences downstream. Both seem to be used to discriminate AAUAAA poly(A) signals from ∼1200 such hexamers in coding sequences. Nonetheless, in Metamonada, irrespective of the poly(A) signal used, known auxiliary elements are neither enriched nor contribute to recognition based on our modeling. Given that these elements play a role in T. brucei (in the Discoba clade), A. thaliana, and H. sapiens, we propose that they were present in the eukaryotic ancestral recognition and then lost early in Metamonada evolution.
In fact, one surprise of our analysis is the diversity of strategies to distinguish premature poly(A) sites from mature ones aside from the previously described upstream and downstream motifs. Our analysis revealed at least three other strategies. One strategy is the identity of the poly(A) signal itself. The best example of this strategy is N. gruberi, although this specification also occurs in G. lamblia. Another strategy is the widespread depletion of poly(A) signals from coding sequences. This strategy seems to be very prevalent in G. muris, but not G. lamblia, indicating a recent, widespread genomic adaptation.
Finally, all studied Metamonada species rely, to some extent, on the nucleotides flanking the poly(A) signal. To our knowledge, a role for these flanking nucleotides—to make a functional octameric poly(A) signal—has not been observed in any species to date. The role of the flanking nucleotides, especially the nucleotide directly downstream from the WGURAA hexamer, is especially strong for G. lamblia species, raising the intriguing possibility that increasing reliance on these poly(A) site-adjacent nucleotides may have been an alternative “strategy” to adapt to the change in poly(A) signal. These flanking nucleotides contribute to recognition in all Metamonada species, including T. vaginalis and T. foetus, which use the ancestral signal. However, weaker model performance for these two species together with the frequent occurrence of AAUAAA hexamers in coding sequences suggests that unknown additional cis-elements are likely involved in T. vaginalis and T. foetus poly(A) signal recognition.
Given that all of these RNA motifs are recognized by RNA-binding proteins, it is reasonable to predict that these proteins have also changed to recognize the derived poly(A) signals and auxiliary elements. In the case of G. lamblia, the streamlined polyadenylation machinery has only six to seven homologs to the human core complex (Ospina-Villa et al. 2020; Bilodeau et al. 2022). The CPSF30 homolog, which is directly involved in poly(A) signal recognition, contains Giardia-specific amino acid changes within two critical zinc finger motifs that are responsible for the recognition of the second and fourth nucleotide in the poly(A) signal (Ospina-Villa et al. 2020; Bilodeau et al. 2022). It is tempting to hypothesize that these mutations may provide a molecular explanation for the difference in poly(A) signals. Similarly, we previously found a putative WDR33 homolog (Bilodeau et al. 2022); this protein also contains amino acid substitutions specific to the Giardia genus, which may have augmented the role of the downstream nucleotide in poly(A) signal recognition. Understanding the structural basis of the so-called octameric poly(A) signal will be an important next step.
Our study thus highlights a hitherto unknown plasticity associated with pre-mRNA CPA and shows the power of exploring fundamental molecular processes beyond the standard model systems. We highlight how new methods like long-read sequencing can improve our understanding of RNA biology across the eukaryotic tree of life and motivate future studies into nontraditional organisms, especially eukaryotic pathogens. In addition to enabling molecular biologic studies (e.g., through the creation of robust transgenic gene expression cassettes), our results set the stage for future structural and molecular studies to better understand interactions between cis-elements and trans factors within protists. Our discoveries highlight a potential avenue for pharmacologically targeting eukaryotic pathogens by virtue of the differences in poly(A) site recognition compared to metazoans.
Methods
G. lamblia (assemblage A, strain WB clone C6) (ATCC 50803) and G. lamblia (assemblage B, strain GS/M, clone H7) (ATCC 50581) were grown in Keister's modified TYI-S-33 medium (Keister 1983). E. histolytica strain HM1-IMSS (ATCC 30459) was grown in TYI-S-33 medium (Diamond et al. 1978). T. foetus strain D1 and T. vaginalis strain F1623 were grown in trypticase–yeast extract–maltose (TYM) medium, supplemented with 10% horse serum (Omega DH-05), 0.74% ammonium iron (II) sulfate hexahydrate (Sigma-Aldrich 215406) and 1% penicillin–streptomycin (Sigma-Aldrich P4333), adjusted to pH 6.2. G. muris Roberts-Thomson isolate was obtained from Waterborne Inc. (P105). Cysts were incubated in 15 mL of the induction medium (0.1 M potassium phosphate buffer pH 7, 0.3 M sodium bicarbonate) in capped centrifuge tubes at 37°C for 10 min (0.5–1.0 × 106 cysts/tube). Samples were then centrifuged at 600g for 5 min, washed in 15 mL of 0.1 M potassium phosphate buffer (pH 7.0), concentrated by centrifugation for 5 min at 600g, and suspended in 1–2 mL of TYI-S-33 medium. All protists were cultured at 37°C. N. gruberi strain NEG-M (ATCC 30224) was obtained from Chandler Fulton at Brandeis University and were grown in M7 medium (0.362 g/L KH2PO4, 0.5 g/L Na2HPO4, 5.4 g/L glucose, 5 g/L yeast extract [Difco], 45 mg/L l-methionine, 10% fetal bovine serum) at 26°C without shaking in 25 cm2 plug-seal tissue culture flasks (CellTreat 229330).
RNA extraction
G. lamblia, E. histolytica, T. vaginalis, and T. foetus were grown in log phase to no more than 80% confluence, detached by cooling on ice (E. histolytica and G. lamblia) or by vigorous shaking (T. vaginalis), and washed twice by centrifugation and resuspension in ice-cold PBS.
G. lamblia assemblage A RNA was isolated using hot acid phenol as previously described (Collart and Oliviero 2001). For remaining protists, the washed pellets were lysed in TRI Reagent (Zymo Research) and RNA was extracted and purified using Direct-zol RNA Miniprep Kit (Zymo Research) following the manufacturer's instructions. G. muris samples were centrifuged (1000g for 5 min), washed twice with PBS, resuspended in 1 mL of TRIzol, and frozen at −20°C. The following day, RNA was isolated following the manufacturer's protocol. Total RNA concentration and purity (A260/A280 ratio) were quantified using a NanoDropTM 1000 instrument (Thermo Fisher Scientific). RNA concentrations were >100 ng/µL and A260/A280 ratios >1.75 for all samples. N. gruberi samples were washed once in 2 mM Tris and centrifuged at 1500g at room temperature. The pellet was suspended in 1 mL TRIzol, vortexed, and stored at −80°C until RNA extraction. Samples were lysed using FastPrep homogenizer with bead beating in TRIzol. Lysate was cleaned up using a Zymo kit with on-column DNase treatment. The RNA concentration and integrity were measured by Qubit (Thermo Fisher Scientific). Concentration was >350 ng/µL and the RIN value was between 5.8 and 6.6. Cell harvesting and RNA extraction were repeated for each parasite strain on three different days.
RNA sequencing and analysis
Nanopore libraries were prepared as described in Bilodeau et al. (2022). Libraries were sequenced on a FLO-MIN106 flow cell and minION sequencing device. For G. lamblia assemblage A, we prepared one new library and used it as a third replicate. Raw data for the other two replicates were from our previous study (Bilodeau et al. 2022). Base-calling was performed using guppy v. 5.0.11 + 2b6dbffa5. Reads were aligned using minimap2 (Li 2018) v. 2.17-r941 with the following parameters: “-a -x splice -uf -k 14”. The genomic annotations used for the analyses are found in Table 1.
Versions and sources of genomes used in the study
| Organism | Genome | Source |
|---|---|---|
| Entamoeba histolytica | Entamoeba_histolytica.JCVI-ESG2-1.0.52 | Ensembl |
| Giardia lamblia A | GiardiaDB-56_GintestinalisAssemblageAWB | VEuPathDB |
| Giardia lamblia B | GiardiaDB-56_GintestinalisAssemblageBGS | VEuPathDB |
| Giardia muris | GiardiaDB-56_GmurisRobertsThomson | VEuPathDB |
| Naegleria gruberi | Naegleria_gruberi_gca_000004985.V1.0.52 | Ensembl |
| Trichomonas vaginalis | TrichDB-56_TvaginalisG3 | VEuPathDB |
| Tritrichomonas foetus | TrichDB-56_TfoetusK | VEuPathDB |
Poly(A) tail length was estimated using nanopolish (Loman et al. 2015) v. 0.13.2. Mapped nanopore reads were assigned to their corresponding genes using featureCounts (Liao et al. 2014) v. 2.8.2 from Rsubread (Liao et al. 2019) using R (R Core Team 2021).
Cleavage sites identification and 3′-UTR annotation
To annotate the cleavage sites, first, we select the reads with poly(A) tail length ≥15 (N. gruberi) or ≥30 (other protists) and for which the 5′ end of the read fell within the open reading frame of the associated gene. Then we selected genes with at least 10 reads total and grouped them by a read start position (3′ end). Reads within the 20 nt window were grouped together as a putative 3′ end if the group contained at least 10% of total reads for a particular gene. Cleavage site was annotated at the 3′ end of the group. If more than one group was present for the gene they were considered 3′-UTR isoforms with alternative CPA sites. The analysis was performed using custom R scripts (Supplemental Code).
Poly(A) signals, “dual use” signals, and auxiliary elements identification and quantification
To identify putative poly(A) signals, we performed de novo motif discovery on the 40 nt upstream of the annotated cleavage sites using MEME (Bailey and Elkan 1994) v. 5.0.5 with the following parameters -rna -mod zoops -minw 3 -maxw 10, and we selected the top motif based on the number of occurrences. MEME PWMs were used to select top 6-mers for all organisms and calculate their frequencies within 40 nt upstream of the annotated cleavage sites.
To identify putative “dual use” signals where the stop codon overlaps with poly(A) signal, we extracted stop codons coordinates for all annotated protein-coding genes in each organism. To capture all possible overlaps, we extended these coordinates 2 nt upstream and 5 nt downstream and searched for the sequences listed in Figure 2A.
Occurrences of UGUA upstream auxiliary element and shuffled versions of this tetramer were calculated in the region 20–50 nt upstream of the cleavage site. The statistical significance of the differences between humans and protists was checked using the χ2 test. Occurrences of GU and U-rich auxiliary elements (CUGCCU, CUGGGG, CUGUGU, GUCUGU, GUGUCU, GUGUGU, UGUCUC, UGUCUG, UGUGUG, UGUUUU, UUAUUU, UUUCUU, UUUUUU) were compared 40 nt downstream from the cleavage versus 40 nt upstream to the cleavage site region. Statistical significance was calculated in comparison to human enrichment using a one-sided Wilcoxon rank sum test. The analyses were performed using custom R scripts (Supplemental Code).
Dicodons occurrence quantification
All in-frame codon combinations encoding Ser-Lys, Lys-Ser, Ser-Cys, Cys-Ser, Ser-Glu, and Glu-Ser were counted within annotated coding sequences for every organism using oligonucleotideFrequency function from Biostrings v. 2.62 R library with the following settings: width = 6, step = 3.
Gapped k-mer support vector machine analysis
The positive and negative sets were prepared as follows: Every AGUAAA|UGUAAA|AGUGAA (G. lamblia, G. muris) or AAUAAA|AUUAAA (N. gruberi, T. vaginalis, T. foetus) hexamer was extracted from either 3′ UTR (positive set) or coding sequence (negative set) and extended 37 nt upstream and 37 nt downstream (total length 80 nt); 80-mers were used to train gkmSVM classifier as described in https://www.beerlab.org/gkmsvm/gkmsvm-tutorial.htm. We used gkmSVM (Ghandi et al. 2014, 2016) v. 0.81 R library with the following settings: k-mer length: 8 (8, 10, and 12 were tested for G. lamblia and G. muris) (Supplemental Table S2), number of informative columns: 6, max number of mismatches: 3, add reverse complement: FALSE. For G. lamblia and G. muris data sets, all positive and negative 80-mers were used for training. For N. gruberi, T. vaginalis, and T. foetus, negative sets were split into five subsets because of sample imbalance, and training was repeated for each subset. Model performance was measured using caret (Kuhn 2008) v. 6.0.92 R library and F1 metrics (Supplemental Table S2). N. gruberi, T. vaginalis, and T. foetus models trained on negative subsets 1 were used for further analysis. The gkmSVM models were used to score all possible 8-mers containing either AGUAAA|UGUAAA|AGUGAA or AAUAAA|AUUAAA. Scores were then used to build linear models with three independent variables: upstream nucleotide, hexamer sequence, and downstream nucleotide (lm R function). To predict putative new auxiliary elements de novo PWMs were built by systematically merging the most predictive k-mers from trained gkmSVM models, as described in Ghandi et al. (2014). K-mers clustering for N. gruberi AAUAAA-containing sequences was performed using kmer v. 1.1.2 R library.
Premature cleavage identification
Coding sequences containing WGURAA hexamers misclassified as 3′ UTRs (G. lamblia A, n = 23, G. lamblia B, n = 14) or all coding sequences containing WGURAA hexamer (G. muris, n = 199) were visually inspected in Integrative Genomics Viewer (Robinson et al. 2011) v. 2.14. If according to the investigator assessment gene was correctly annotated with a substantial coverage drop within 20 nt downstream from WGURAA hexamer, it was defined as premature cleavage event. Coverage data for genes selected to figure preparation was converted from BAM to bigWig format using deepTools (Ramírez et al. 2014) v. 3.5.1 and visualized using ggcoverage (Song and Wang 2023) v. 1.2.0 R library. Duplication events for genes undergoing premature cleavage were identified using OrthoFinder (Emms and Kelly 2019) v. 2.5.4.
Data analysis, statistical tests, and visualization
Statistical details of specific experiments can be found in the Results, Methods, and/or Figure Legends. Raw plots were generated using the R ggplot2 (Wickham 2016) or ggpubr (Kassambara 2019) packages. Final figures were generated using Adobe Illustrator.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE260731. All code used for data analysis and figure generation is accessible at GitHub (https://github.com/mukherjeelab/2024_protists_polyA) and as Supplemental Code.
Competing interest statement
O.S.R. is a member of the Cell Reports and Molecular Cell Scientific Advisory Boards.
Acknowledgments
We thank J. Mathew Taliaferro, David Bentley, and Srinivas Ramachandran for the critical comments. This work was supported by the Polish National Agency for Academic Exchange Bekker Program PPN/BEK/2019/1/00173 (M.P.S.), the University of Colorado Anschutz Medical Campus RNA Bioscience Initiative (M.P.S., N.M., and O.S.R.), the National Institute of Health grants R01 AI158612 (L.E.), P30 DK120515 (L.E.), R35 GM128680 (O.S.R.), R35 GM147025 (N.M.), and a Pew Biomedical Scholar Award to L.F.-L. who is a fellow in the Canadian Institute for Advanced Research Fungal Kingdom program.
Author contributions: O.S.R., N.M., and M.P.S.: conceptualization; M.P.S., M.A.B., N.M., and O.S.R.: methodology; M.P.S. and M.A.B.: software; M.P.S. and M.A.B.: formal analysis; M.P.S., D.Y.B., N.M., and O.S.R.: investigation; Y.M., K.B.V., L.E., L.F.-L., N.M., and O.S.R.: resources; M.P.S.: data curation; M.P.S.: writing—original draft; M.P.S., N.M., and O.S.R.: writing—review and editing; M.P.S., E.H., N.M., and O.S.R.: visualization; N.M. and O.S.R.: supervision; N.M. and O.S.R.: project administration; M.P.S., L.E., L.F.-L., N.M., and O.S.R.: funding acquisition.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279526.124.
References
- ↵Adam RD, Dahlstrom EW, Martens CA, Bruno DP, Barbian KD, Ricklefs SM, Hernandez MM, Narla NP, Patel RB, Porcella SF, 2013. Genome sequencing of Giardia lamblia genotypes A2 and B isolates (DH and GS) and comparative analysis with the genomes of genotypes A1 and E (WB and Pig). Genome Biol Evol 5: 2498–2511. 10.1093/gbe/evt197
- ↵Baer BW, Kornberg RD. 1983. The protein responsible for the repeating structure of cytoplasmic poly(A)-ribonucleoprotein. J Cell Biol 96: 717–721. 10.1083/jcb.96.3.717
- ↵Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36.
- ↵Beyer K, Dandekar T, Keller W. 1997. RNA ligands selected by cleavage stimulation factor contain distinct sequence motifs that function as downstream elements in 3′-end processing of pre-mRNA. J Biol Chem 272: 26769–26779. 10.1074/jbc.272.42.26769
- ↵Bilodeau DY, Sheridan RM, Balan B, Jex AR, Rissland OS. 2022. Precise gene models using long-read sequencing reveal a unique poly(A) signal in Giardia lamblia. RNA 28: 668–682. 10.1261/rna.078793.121
- ↵Birse CE. 1997. Transcriptional termination signals for RNA polymerase II in fission yeast. EMBO J 16: 3633–3643. 10.1093/emboj/16.12.3633
- ↵Brown KM, Gilmartin GM. 2003. A mechanism for the regulation of pre-mRNA 3′ processing by human cleavage factor Im. Mol Cell 12: 1467–1476. 10.1016/s1097-2765(03)00453-2
- ↵Chan S, Choi E-A, Shi Y. 2011. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip Rev RNA 2: 321–335. 10.1002/wrna.54
- ↵Chen F, MacDonald CC, Wilusf J. 1995. Cleavage site determinants in the mammalian polyadenylation signal. Nucleic Acids Res 23: 2614–2620. 10.1093/nar/23.14.2614
- ↵Collart MA, Oliviero S. 2001. Preparation of yeast RNA. Curr Protoc Mol Biol Chapter 13: Unit13.12. 10.1002/0471142727.mb1312s23
- ↵Dávila López M, Samuelsson T. 2008. Early evolution of histone mRNA 3′ end processing. RNA 14: 1–10. 10.1261/rna.782308
- ↵Diamond LS, Harlow DR, Cunnick CC. 1978. A new medium for the axenic cultivation of Entamoeba histolytica and other Entamoeba. Trans R Soc Trop Med Hyg 72: 431–432. 10.1016/0035-9203(78)90144-x
- ↵Emms DM, Kelly S. 2019. Orthofinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20: 238. 10.1186/s13059-019-1832-y
- ↵Espinosa N, Hernández R, López-Griego L, López-Villaseñor I. 2002. Separable putative polyadenylation and cleavage motifs in Trichomonas vaginalis mRNAs. Gene 289: 81–86. 10.1016/s0378-1119(02)00476-6
- ↵Fitzgerald M, Shenk T. 1981. The sequence 5′-AAUAAA-3′ forms parts of the recognition site for polyadenylation of late SV40 mRNAs. Cell 24: 251–260. 10.1016/0092-8674(81)90521-3
- ↵Franzén O, Jerlström-Hultqvist J, Einarsson E, Ankarklev J, Ferella M, Andersson B, Svärd SG. 2013. Transcriptome profiling of Giardia intestinalis using strand-specific RNA-seq. PLoS Comput Biol 9: e1003000. 10.1371/journal.pcbi.1003000
- ↵Fuentes V, Barrera G, Sánchez J, Hernández R, López-Villaseñor I. 2012. Functional analysis of sequence motifs involved in the polyadenylation of Trichomonas vaginalis mRNAs. Eukaryot Cell 11: 725–734. 10.1128/ec.05322-11
- ↵Ghandi M, Lee D, Mohammad-Noori M, Beer MA. 2014. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput Biol 10: e1003711. 10.1371/journal.pcbi.1003711
- ↵Ghandi M, Mohammad-Noori M, Ghareghani N, Lee D, Garraway L, Beer MA. 2016. gkmSVM: an R package for gapped-kmer SVM. Bioinformatics 32: 2205–2207. 10.1093/bioinformatics/btw203
- ↵Graber JH, Cantor CR, Mohr SC, Smith TF. 1999a. Genomic detection of new yeast pre-mRNA 3′-end-processing signals. Nucleic Acids Res 27: 888–894. 10.1093/nar/27.3.888
- ↵Graber JH, Cantor CR, Mohr SC, Smith TF. 1999b. In silico detection of control signals: mRNA 3′-end-processing sequences in diverse species. Proc Natl Acad Sci 96: 14055–14060. 10.1073/pnas.96.24.14055
- ↵Guo Z, Sherman F. 1996. Signals sufficient for 3′-end formation of yeast mRNA. Mol Cell Biol 16: 2772–2776. 10.1128/MCB.16.6.2772
- ↵Higgs DR, Goodbourn SE, Lamb J, Clegg JB, Weatherall DJ, Proudfoot NJ. 1983. Alpha-thalassaemia caused by a polyadenylation signal mutation. Nature 306: 398–400. 10.1038/306398a0
- ↵Hon C-C, Weber C, Sismeiro O, Proux C, Koutero M, Deloger M, Das S, Agrahari M, Dillies M-A, Jagla B, 2013. Quantification of stochastic noise of splicing and polyadenylation in Entamoeba histolytica. Nucleic Acids Res 41: 1936–1952. 10.1093/nar/gks1271
- ↵Hu J, Lutz CS, Wilusz J, Tian B. 2005. Bioinformatic identification of candidate cis-regulatory elements involved in human mRNA polyadenylation. RNA 11: 1485–1493. 10.1261/rna.2107305
- ↵Huang K-Y, Shin J-W, Huang P-J, Ku F-M, Lin W-C, Lin R, Hsu W-M, Tang P. 2013. Functional profiling of the Tritrichomonas foetus transcriptome and proteome. Mol Biochem Parasitol 187: 60–71. 10.1016/j.molbiopara.2012.12.001
- ↵Kassambara A. 2019. GGPlot2 essentials. Independently Published.
- ↵Keister DB. 1983. Axenic culture of Giardia lamblia in TYI-S-33 medium supplemented with bile. Trans R Soc Trop Med Hyg 77: 487–488. 10.1016/0035-9203(83)90120-7
- ↵Kuhn M. 2008. Building predictive models in R using the caret package. J Stat Softw 28: 1–26. 10.18637/jss.v028.i05
- ↵Kumar A, Clerici M, Muckenfuss LM, Passmore LA, Jinek M. 2019. Mechanistic insights into mRNA 3′-end processing. Curr Opin Struct Biol 59: 143–150. 10.1016/j.sbi.2019.08.001
- ↵Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191
- ↵Li X-Q, Du D. 2014. Motif types, motif locations and base composition patterns around the RNA polyadenylation site in microorganisms, plants and animals. BMC Evol Biol 14: 162. 10.1186/s12862-014-0162-7
- ↵Li Q, Hunt AG. 1997. The polyadenylation of RNA in plants. Plant Physiol 115: 321–325. 10.1104/pp.115.2.321
- ↵Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. 10.1093/bioinformatics/btt656
- ↵Liao Y, Smyth GK, Shi W. 2019. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res 47: e47. 10.1093/nar/gkz114
- ↵Liu X, Hoque M, Larochelle M, Lemay J-F, Yurko N, Manley JL, Bachand F, Tian B. 2017. Comparative analysis of alternative polyadenylation in S. cerevisiae and S. pombe. Genome Res 27: 1685–1695. 10.1101/gr.222331.117
- ↵Loman NJ, Quick J, Simpson JT. 2015. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 12: 733–735. 10.1038/nmeth.3444
- ↵Mandel CR, Kaneko S, Zhang H, Gebauer D, Vethantham V, Manley JL, Tong L. 2006. Polyadenylation factor CPSF-73 is the pre-mRNA 3′-end-processing endonuclease. Nature 444: 953–956. 10.1038/nature05363
- ↵Mandel CR, Bai Y, Tong L. 2008. Protein factors in pre-mRNA 3′-end processing. Cell Mol Life Sci 65: 1099–1122. 10.1007/s00018-007-7474-3
- ↵Montell C, Fisher EF, Caruthers MH, Berk AJ. 1983. Inhibition of RNA cleavage but not polyadenylation by a point mutation in mRNA 3′ consensus sequence AAUAAA. Nature 305: 600–605. 10.1038/305600a0
- ↵Ospina-Villa JD, Tovar-Ayona BJ, López-Camarillo C, Soto-Sánchez J, Ramírez-Moreno E, Castañón-Sánchez CA, Marchat LA. 2020. MRNA polyadenylation machineries in intestinal protozoan parasites. J Eukaryot Microbiol 67: 306–320. 10.1111/jeu.12781
- ↵Proudfoot NJ, Brownlee GG. 1976. 3′ non-coding region sequences in eukaryotic messenger RNA. Nature 263: 211–214. 10.1038/263211a0
- ↵Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. 2014. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42: W187–W191. 10.1093/nar/gku365
- ↵R Core Team. 2021. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna https://www.R-project.org/.
- ↵Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754
- ↵Rothnie HM. 1996. Plant mRNA 3′-end formation. Plant Mol Biol 32: 43–61. 10.1007/BF00039376
- ↵Sheets MD, Ogg SC, Wickens MP. 1990. Point mutations in AAUAAA and the poly (A) addition site: effects on the accuracy and efficiency of cleavage and polyadenylation in vitro. Nucleic Acids Res 18: 5799–5805. 10.1093/nar/18.19.5799
- ↵Shen Y, Liu Y, Liu L, Liang C, Li QQ. 2008. Unique features of nuclear mRNA poly(A) signals and alternative polyadenylation in Chlamydomonas reinhardtii. Genetics 179: 167–176. 10.1534/genetics.108.088971
- ↵Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JRIII, Frank J, Manley JL. 2009. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol Cell 33: 365–376. 10.1016/j.molcel.2008.12.028
- ↵Song Y, Wang J. 2023. ggcoverage: an R package to visualize and annotate genome coverage for various NGS data. BMC Bioinformatics 24: 309. 10.1186/s12859-023-05438-2
- ↵Sullivan KD, Steiniger M, Marzluff WF. 2009. A core complex of CPSF73, CPSF100, and Symplekin may form two different cleavage factors for processing of poly(A) and histone mRNAs. Mol Cell 34: 322–332. 10.1016/j.molcel.2009.04.024
- ↵Takagaki Y, Manley JL. 1997. RNA recognition by the human polyadenylation factor CstF. Mol Cell Biol 17: 3907–3914. 10.1128/MCB.17.7.3907
- ↵Takyar S, Hickerson RP, Noller HF. 2005. mRNA helicase activity of the ribosome. Cell 120: 49–58. 10.1016/j.cell.2004.11.042
- ↵Venkataraman K, Brown KM, Gilmartin GM. 2005. Analysis of a noncanonical poly(A) site reveals a tripartite mechanism for vertebrate poly(A) site recognition. Genes Dev 19: 1315–1327. 10.1101/gad.1298605
- ↵Wickham H. 2016. ggplot2, 2nd ed. Springer International Publishing, Basel, Switzerland.
- ↵Wu L, Ueda T, Messing J. 1995. The formation of mRNA 3′-ends in plants. Plant J 8: 323–329. 10.1046/j.1365-313x.1995.08030323.x
- ↵Xu F, Jerlström-Hultqvist J, Einarsson E, Ástvaldsson Á, Svärd SG, Andersson JO. 2014. The genome of Spironucleus salmonicida highlights a fish pathogen adapted to fluctuating environments. PLoS Genet 10: e1004053. 10.1371/journal.pgen.1004053
- ↵Xu F, Jiménez-González A, Einarsson E, Ástvaldsson Á, Peirasmaki D, Eckmann L, Andersson JO, Svärd SG, Jerlström-Hultqvist J. 2020. The compact genome of Giardia muris reveals important steps in the evolution of intestinal protozoan parasites. Microb Genom 6: mgen000402. 10.1099/mgen.0.000402
- ↵Yee J, Tang A, Lau W-L, Ritter H, Delport D, Page M, Adam RD, Müller M, Wu G. 2007. Core histone genes of Giardia intestinalis: genomic organization, promoter structure, and expression. BMC Mol Biol 8: 26. 10.1186/1471-2199-8-26
- ↵Yusupova GZ, Yusupov MM, Cate JH, Noller HF. 2001. The path of messenger RNA through the ribosome. Cell 106: 233–241. 10.1016/s0092-8674(01)00435-4
- ↵Zajaczkowski P, Lee R, Fletcher-Lartey SM, Alexander K, Mahimbo A, Stark D, Ellis JT. 2021. The controversies surrounding Giardia intestinalis assemblages A and B. Curr Res Parasitol Vector Borne Dis 1: 100055. 10.1016/j.crpvbd.2021.100055
- ↵Zarudnaya MI, Kolomiets IM, Potyahaylo AL, Hovorun DM. 2003. Downstream elements of mammalian pre-mRNA polyadenylation signals: primary, secondary and higher-order structures. Nucleic Acids Res 31: 1375–1386. 10.1093/nar/gkg241
- ↵Zhao Z, Wu X, Ji G, Liang C, Li QQ. 2019. Genome-wide comparative analyses of polyadenylation signals in eukaryotes suggest a possible origin of the AAUAAA signal. Int J Mol Sci 20: 958. 10.3390/ijms20040958